Несмотря на то,
что термодинамика не учитывает процессы,
происходящие в реальных растворах,
например, притяжение и отталкивание
ионов, термодинамические закономерности,
выведенные для идеальных растворов,
можно применить и для реальных растворов,
если заменить концентрации активностями.
Активность (a) -такая концентрация вещества в растворе, при использовании которой свойства данного раствора могут быть описаны теми же уравнениями, что и свойства идеального раствора.
Активность может
быть как меньше, так и больше номинальной
концентрации вещества в растворе.
Активность чистого растворителя, а
также растворителя в не слишком
концентрированных растворах принимается
равной 1. За 1 принимается также активность
твёрдого вещества, находящегося в
осадке, или жидкости, не смешивающейся
с данным раствором. В бесконечно
разбавленном растворе активность
растворённого вещества совпадает с его
концентрацией.
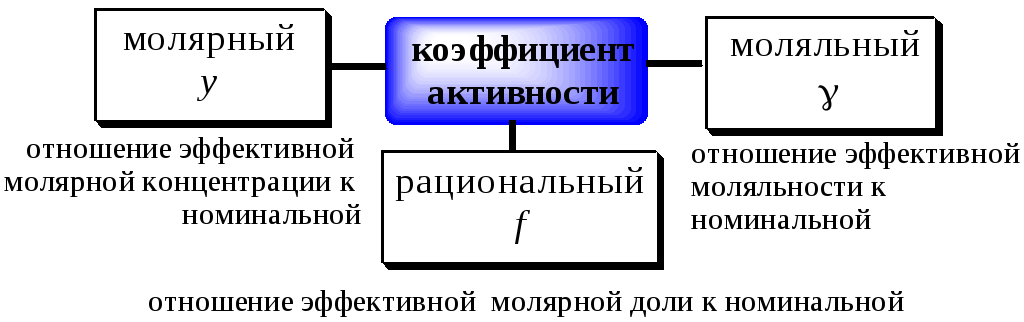
Отношение
активности вещества в данном растворе
к его концентрации называетсякоэффициентом активности.
Коэффициент
активности — это своеобразный поправочный
коэффициент, показывающий, насколько
реальность отличается от идеала.
3.3.
Отклонения от идеальности в растворах
сильных электролитов
Особенно заметное
отклонение от идеальности имеет место
в растворах сильных электролитов. Это
отражается, например, на их температурах
кипения, плавления, давлении пара над
раствором и, что особенно важно для
аналитической химии, на величинах
констант различных равновесий, протекающих
в таких растворах.
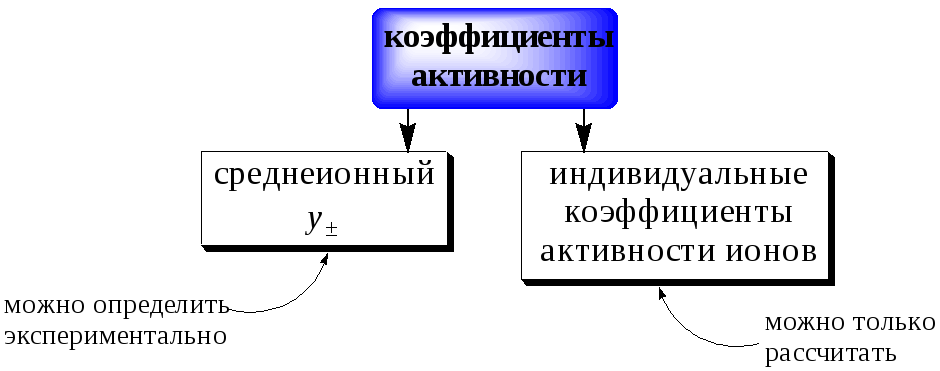
Для характеристики
активности электролитов используют:
Для электролита
AmBn:
![]()
Величина, которая
учитывает влияние концентрации (С) и
заряда (z) всех ионов,
присутствующих в растворе, на активность
растворённого вещества, называетсяионной силой (I).
![]()
Пример
3.1.В 1,00 л водного раствора
содержится 10,3 г NaBr,
14,2 г Na2SO4
и 1,7 г NH3.
Чему равна ионная сила такого раствора?
0,100 Моль/л
0,100 Моль/л
С(Na+)
= 0,300 моль/л, С(Br—) =
0,100 моль/л, С(SO42-)
= 0,100моль/л
I=
0,5[0,300(+1)2+ 0,100(-1)2+ 0,100(-2)2]
= 0,400 моль/л
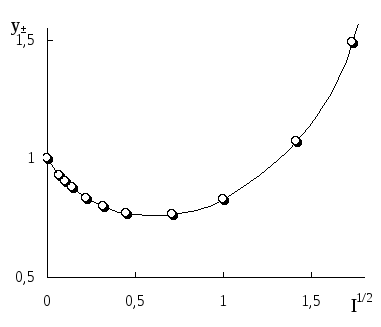
Рис.
3.1. Влияние
ионной силы на среднеионный коэффициент
активности HCl
На рис. 3.1 показан
пример влияния ионной силы на активность
электролита (HCl). Аналогичная
зависимость коэффициента активности
от ионной силы наблюдается также уHClO4,LiCl,AlCl3и многих других
соединений. У некоторых электролитов
(NH4NO3,AgNO3) зависимость
коэффициента активности от ионной силы
является монотонно убывающей.
Универсального
уравнения, с помощью которого можно
было бы рассчитать коэффициент активности
любого электролита при любой величине
ионной силы, не существует. Для описания
зависимости коэффициента активности
от ионной силы в очень разбавленных
растворах (до I< 0,01) можно
использоватьпредельный закон
Дебая-Хюккеля
![]()
где
A
— коэффициент, зависящий от температуры
и диэлектрической проницаемости среды;
для водного раствора (298К) A
0,511.
Данное уравнение
было получено голландским физиком П.
Дебаем и его учеником Э. Хюккелем исходя
из следующих предположений. Каждый ион
был представлен в виде точечного заряда
(т.е. размер иона не учитывался), окружённого
в растворе ионной атмосферой—
областью пространства сферической
формы и определённого размера, в которой
содержание ионов противоположного
знака по отношению к данному иону больше,
чем вне её. Заряд ионной атмосферы равен
по величине и противоположен по знаку
заряду создавшего её центрального иона.
Между центральным ионом и окружающей
его ионной атмосферой существует
электростатическое притяжение, которое
стремится стабилизировать данный ион.
Стабилизация приводит к понижению
свободной энергии иона и уменьшению
его коэффициента активности. В предельном
уравнении Дебая-Хюккеля природа ионов
не учитывается. Считается, что при малых
значениях ионной силы коэффициент
активности иона не зависит от его
природы.
При
увеличении ионной силы до 0,01 и больше
предельный закон начинает давать всё
большую и большую погрешность. Это
происходит потому, что реальные ионы
имеют определённый размер, вследствие
чего их нельзя упаковать так плотно,
как точечные заряды. При увеличении
концентрации ионов происходит уменьшение
размеров ионной атмосферы. Так как
ионная атмосфера стабилизирует ион и
уменьшает его активность, то уменьшение
её размера приводит к менее значительному
уменьшению коэффициента активности.
Для расчёта
коэффициентов активности при ионных
силах порядка 0,01 — 0,1 можно использовать
расширенное уравнение Дебая-Хюккеля:
![]()
где
B
0,328 (T
= 298K,
a
выражено в ),
a
— эмпирическая константа, характеризующая
размеры ионной атмосферы.
При более высоких
значениях ионной силы (до 1)
количественную оценку коэффициента
активности можно проводить поуравнению
Дэвиса.
![]()
В
данном уравнении a
принято равным 3,05, поэтому произведение
Ba
равно 1. Фактор 0,2I
учитывает образование ионных пар,
изменение диэлектрической проницаемости
и т.д.
В ещё более
концентрированных растворах начинают
сильно проявляться индивидуальные
особенности ионов, поэтому уравнения,
описывающего экспериментальные данные
для таких растворов, нет.У
одних электролитов коэффициент активности
уменьшается, что может быть обусловлено
образованием ионных пар, у других он
увеличивается — за счёт уменьшения не
принимающих участие в гидратации молекул
воды и по другим причинам.
Пример
3.2. Рассчитать коэффициенты
активности иона H+
при ионной силе 0,010 и 0,10.
При I
= 0,010 ![]() = -0,0511;
= -0,0511;
![]() 0,89.
0,89.
При I
= 0,10
![]() =-0,0836,
=-0,0836,
![]() =
=
0,82
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
From Wikipedia, the free encyclopedia
In thermodynamics, an activity coefficient is a factor used to account for deviation of a mixture of chemical substances from ideal behaviour.[1] In an ideal mixture, the microscopic interactions between each pair of chemical species are the same (or macroscopically equivalent, the enthalpy change of solution and volume variation in mixing is zero) and, as a result, properties of the mixtures can be expressed directly in terms of simple concentrations or partial pressures of the substances present e.g. Raoult’s law. Deviations from ideality are accommodated by modifying the concentration by an activity coefficient. Analogously, expressions involving gases can be adjusted for non-ideality by scaling partial pressures by a fugacity coefficient.
The concept of activity coefficient is closely linked to that of activity in chemistry.
Thermodynamic definition[edit]
The chemical potential,  , of a substance B in an ideal mixture of liquids or an ideal solution is given by
, of a substance B in an ideal mixture of liquids or an ideal solution is given by
 ,
,

where μo
B is the chemical potential of a pure substance  , and
, and  is the mole fraction of the substance in the mixture.
is the mole fraction of the substance in the mixture.
This is generalised to include non-ideal behavior by writing

when  is the activity of the substance in the mixture,
is the activity of the substance in the mixture,
- ,

where  is the activity coefficient, which may itself depend on . As approaches 1, the substance behaves as if it were ideal. For instance, if ≈ 1, then Raoult’s law is accurate. For > 1 and < 1, substance B shows positive and negative deviation from Raoult’s law, respectively. A positive deviation implies that substance B is more volatile.
is the activity coefficient, which may itself depend on . As approaches 1, the substance behaves as if it were ideal. For instance, if ≈ 1, then Raoult’s law is accurate. For > 1 and < 1, substance B shows positive and negative deviation from Raoult’s law, respectively. A positive deviation implies that substance B is more volatile.
In many cases, as goes to zero, the activity coefficient of substance B approaches a constant; this relationship is Henry’s law for the solvent. These relationships are related to each other through the Gibbs–Duhem equation.[2]
Note that in general activity coefficients are dimensionless.
In detail: Raoult’s law states that the partial pressure of component B is related to its vapor pressure (saturation pressure) and its mole fraction in the liquid phase,

with the convention

In other words: Pure liquids represent the ideal case.
At infinite dilution, the activity coefficient approaches its limiting value, ∞. Comparison with Henry’s law,

immediately gives

In other words: The compound shows nonideal behavior in the dilute case.
The above definition of the activity coefficient is impractical if the compound does not exist as a pure liquid. This is often the case for electrolytes or biochemical compounds. In such cases, a different definition is used that considers infinite dilution as the ideal state:

with

and

The  symbol has been used here to distinguish between the two kinds of activity coefficients. Usually it is omitted, as it is clear from the context which kind is meant. But there are cases where both kinds of activity coefficients are needed and may even appear in the same equation, e.g., for solutions of salts in (water + alcohol) mixtures. This is sometimes a source of errors.
symbol has been used here to distinguish between the two kinds of activity coefficients. Usually it is omitted, as it is clear from the context which kind is meant. But there are cases where both kinds of activity coefficients are needed and may even appear in the same equation, e.g., for solutions of salts in (water + alcohol) mixtures. This is sometimes a source of errors.
Modifying mole fractions or concentrations by activity coefficients gives the effective activities of the components, and hence allows expressions such as Raoult’s law and equilibrium constants to be applied to both ideal and non-ideal mixtures.
Knowledge of activity coefficients is particularly important in the context of electrochemistry since the behaviour of electrolyte solutions is often far from ideal, due to the effects of the ionic atmosphere. Additionally, they are particularly important in the context of soil chemistry due to the low volumes of solvent and, consequently, the high concentration of electrolytes.[3]
Ionic solutions[edit]
For solution of substances which ionize in solution the activity coefficients of the cation and anion cannot be experimentally determined independently of each other because solution properties depend on both ions. Single ion activity coefficients must be linked to the activity coefficient of the dissolved electrolyte as if undissociated. In this case a mean stoichiometric activity coefficient of the dissolved electrolyte, γ±, is used. It is called stoichiometric because it expresses both the deviation from the ideality of the solution and the incomplete ionic dissociation of the ionic compound which occurs especially with the increase of its concentration.
For a 1:1 electrolyte, such as NaCl it is given by the following:

where  and
and  are the activity coefficients of the cation and anion respectively.
are the activity coefficients of the cation and anion respectively.
More generally, the mean activity coefficient of a compound of formula  is given by[4]
is given by[4]
![{displaystyle gamma _{pm }={sqrt[{p+q}]{gamma _{mathrm {A} }^{p}gamma _{mathrm {B} }^{q}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c66f131bcf882828c4edbb068ee7800812be4e47)
Single-ion activity coefficients can be calculated theoretically, for example by using the Debye–Hückel equation. The theoretical equation can be tested by combining the calculated single-ion activity coefficients to give mean values which can be compared to experimental values.
The prevailing view that single ion activity coefficients are unmeasurable independently, or perhaps even physically meaningless, has its roots in the work of Guggenheim in the late 1920s.[5] However, chemists have never been able to give up the idea of single ion activities, and by implication single ion activity coefficients. For example, pH is defined as the negative logarithm of the hydrogen ion activity. If the prevailing view on the physical meaning and measurability of single ion activities is correct then defining pH as the negative logarithm of the hydrogen ion activity places the quantity squarely in the unmeasurable category. Recognizing this logical difficulty, International Union of Pure and Applied Chemistry (IUPAC) states that the activity-based definition of pH is a notional definition only.[6] Despite the prevailing negative view on the measurability of single ion coefficients, the concept of single ion activities continues to be discussed in the literature, and at least one author presents a definition of single ion activity in terms of purely thermodynamic quantities and proposes a method of measuring single ion activity coefficients based on purely thermodynamic processes.[7]
Concentrated ionic solutions[edit]
For concentrated ionic solutions the hydration of ions must be taken into consideration, as done by Stokes and Robinson in their hydration model from 1948.[8] The activity coefficient of the electrolyte is split into electric and statistical components by E. Glueckauf who modifies the Robinson–Stokes model.
The statistical part includes hydration index number h, the number of ions from the dissociation and the ratio r between the apparent molar volume of the electrolyte and the molar volume of water and molality b.
Concentrated solution statistical part of the activity coefficient is:
- [9][10][11]

The Stokes–Robinson model has been analyzed and improved by other investigators as well.[12][13]
Experimental determination of activity coefficients[edit]
Activity coefficients may be determined experimentally by making measurements on non-ideal mixtures. Use may be made of Raoult’s law or Henry’s law to provide a value for an ideal mixture against which the experimental value may be compared to obtain the activity coefficient. Other colligative properties, such as osmotic pressure may also be used.
Radiochemical methods[edit]
Activity coefficients can be determined by radiochemical methods.[14]
At infinite dilution[edit]
Activity coefficients for binary mixtures are often reported at the infinite dilution of each component. Because activity coefficient models simplify at infinite dilution, such empirical values can be used to estimate interaction energies. Examples are given for water:
| X | γx∞ (K) | γW∞ (K) |
|---|---|---|
| Ethanol | 4.3800 (283.15) | 3.2800 (298.15) |
| Acetone | 6.0200 (307.85) |
Theoretical calculation of activity coefficients[edit]

Activity coefficients of electrolyte solutions may be calculated theoretically, using the Debye–Hückel equation or extensions such as the Davies equation,[16] Pitzer equations[17] or TCPC model.[18][19][20][21] Specific ion interaction theory (SIT)[22] may also be used.
For non-electrolyte solutions correlative methods such as UNIQUAC, NRTL, MOSCED or UNIFAC may be employed, provided fitted component-specific or model parameters are available. COSMO-RS is a theoretical method which is less dependent on model parameters as required information is obtained from quantum mechanics calculations specific to each molecule (sigma profiles) combined with a statistical thermodynamics treatment of surface segments.[23]
For uncharged species, the activity coefficient γ0 mostly follows a salting-out model:[24]

This simple model predicts activities of many species (dissolved undissociated gases such as CO2, H2S, NH3, undissociated acids and bases) to high ionic strengths (up to 5 mol/kg). The value of the constant b for CO2 is 0.11 at 10 °C and 0.20 at 330 °C.[25]
For water as solvent, the activity aw can be calculated using:[24]

where ν is the number of ions produced from the dissociation of one molecule of the dissolved salt, b is the molality of the salt dissolved in water, φ is the osmotic coefficient of water, and the constant 55.51 represents the molality of water. In the above equation, the activity of a solvent (here water) is represented as inversely proportional to the number of particles of salt versus that of the solvent.
Link to ionic diameter[edit]
The ionic activity coefficient is connected to the ionic diameter by the formula obtained from Debye–Hückel theory of electrolytes:

where A and B are constants, zi is the valence number of the ion, and I is ionic strength.
Dependence on state parameters[edit]
The derivative of an activity coefficient with respect to temperature is related to excess molar enthalpy by

Similarly, the derivative of an activity coefficient with respect to pressure can be related to excess molar volume.

Application to chemical equilibrium[edit]
At equilibrium, the sum of the chemical potentials of the reactants is equal to the sum of the chemical potentials of the products. The Gibbs free energy change for the reactions, ΔrG, is equal to the difference between these sums and therefore, at equilibrium, is equal to zero. Thus, for an equilibrium such as


Substitute in the expressions for the chemical potential of each reactant:

Upon rearrangement this expression becomes

The sum
σμo
S + τμo
T − αμo
A − βμo
B is the standard free energy change for the reaction,  .
.
Therefore,

where K is the equilibrium constant. Note that activities and equilibrium constants are dimensionless numbers.
This derivation serves two purposes. It shows the relationship between standard free energy change and equilibrium constant. It also shows that an equilibrium constant is defined as a quotient of activities. In practical terms this is inconvenient. When each activity is replaced by the product of a concentration and an activity coefficient, the equilibrium constant is defined as
![{displaystyle K={frac {[mathrm {S} ]^{sigma }[mathrm {T} ]^{tau }}{[mathrm {A} ]^{alpha }[mathrm {B} ]^{beta }}}times {frac {gamma _{mathrm {S} }^{sigma }gamma _{mathrm {T} }^{tau }}{gamma _{mathrm {A} }^{alpha }gamma _{mathrm {B} }^{beta }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/95d7e43431f4648654306641285e434365d7af57)
where [S] denotes the concentration of S, etc. In practice equilibrium constants are determined in a medium such that the quotient of activity coefficient is constant and can be ignored, leading to the usual expression
![{displaystyle K={frac {[mathrm {S} ]^{sigma }[mathrm {T} ]^{tau }}{[mathrm {A} ]^{alpha }[mathrm {B} ]^{beta }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/59e684b2a458ef9a608ef9a8d8f9e55473da1e86)
which applies under the conditions that the activity quotient has a particular (constant) value.
References[edit]
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the «Gold Book») (1997). Online corrected version: (2006–) «Activity coefficient». doi:10.1351/goldbook.A00116
- ^ DeHoff, Robert (2018). «Thermodynamics in materials science». Entropy (2nd ed.). 20 (7): 230–231. Bibcode:2018Entrp..20..532G. doi:10.3390/e20070532. ISBN 9780849340659. PMC 7513056. PMID 33265621.
- ^ Ibáñez, Jorge G.; Hernández Esparza, Margarita; Doría Serrano, Carmen; Singh, Mono Mohan (2007). Environmental Chemistry: Fundamentals. Springer. ISBN 978-0-387-26061-7.
- ^ Atkins, Peter; dePaula, Julio (2006). «Section 5.9, The activities of ions in solution». Physical Chemisrry (8th ed.). OUP. ISBN 9780198700722.
- ^ Guggenheim, E. A. (1928). «The Conceptions of Electrical Potential Difference between Two Phases and the Individual Activities of Ions». The Journal of Physical Chemistry. 33 (6): 842–849. doi:10.1021/j150300a003. ISSN 0092-7325.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the «Gold Book») (1997). Online corrected version: (2006–) «pH». doi:10.1351/goldbook.P04524
- ^ Rockwood, Alan L. (2015). «Meaning and Measurability of Single-Ion Activities, the Thermodynamic Foundations of pH, and the Gibbs Free Energy for the Transfer of Ions between Dissimilar Materials». ChemPhysChem. 16 (9): 1978–1991. doi:10.1002/cphc.201500044. ISSN 1439-4235. PMC 4501315. PMID 25919971.
- ^ Stokes, R. H; Robinson, R. A (1948). «Ionic Hydration and Activity in Electrolyte Solutions». Journal of the American Chemical Society. 70 (5): 1870–1878. doi:10.1021/ja01185a065. PMID 18861802.
- ^ Glueckauf, E. (1955). «The influence of ionic hydration on activity coefficients in concentrated electrolyte solutions». Transactions of the Faraday Society. 51: 1235. doi:10.1039/TF9555101235.
- ^ Glueckauf, E. (1957). «The influence of ionic hydration on activity coefficients in concentrated electrolyte solutions». Transactions of the Faraday Society. 53: 305. doi:10.1039/TF9575300305.
- ^ Kortüm, G. (1959). «The Structure of Electrolytic Solutions». Angewandte Chemie. London: Herausgeg. von W. J. Hamer; John Wiley & Sons, Inc., New York; Chapman & Hall, Ltd. 72 (24): 97. doi:10.1002/ange.19600722427. ISSN 0044-8249.
- ^ Miller, Donald G. (1956). «On the Stokes-Robinson Hydration Model for Solutions». The Journal of Physical Chemistry. 60 (9): 1296–1299. doi:10.1021/j150543a034.
- ^ Nesbitt, H. Wayne (1982). «The stokes and robinson hydration theory: A modification with application to concentrated electrolyte solutions». Journal of Solution Chemistry. 11 (6): 415–422. doi:10.1007/BF00649040. S2CID 94189765.
- ^ Betts, R. H.; MacKenzie, Agnes N. (1952). «Radiochemical Measurements of Activity Coefficients in Mixed Electrolytes». Canadian Journal of Chemistry. 30 (2): 146–162. doi:10.1139/v52-020.
- ^ «Activity Coefficients at Infinite Dilution of 30 Important Components from Dortmund Data Bank». Dortmund Data Bank. DDBST GmbH. Retrieved 13 December 2018.
- ^ King, E. L. (1964). «Book Review: Ion Association, C. W. Davies, Butterworth, Washington, D.C., 1962». Science. 143 (3601): 37. Bibcode:1964Sci…143…37D. doi:10.1126/science.143.3601.37. ISSN 0036-8075.
- ^ Grenthe, I.; Wanner, H. «Guidelines for the extrapolation to zero ionic strength» (PDF). Archived from the original (PDF) on 2008-12-17. Retrieved 2007-07-23.
- ^ Ge, Xinlei; Wang, Xidong; Zhang, Mei; Seetharaman, Seshadri (2007). «Correlation and Prediction of Activity and Osmotic Coefficients of Aqueous Electrolytes at 298.15 K by the Modified TCPC Model». Journal of Chemical & Engineering Data. 52 (2): 538–547. doi:10.1021/je060451k. ISSN 0021-9568.
- ^ Ge, Xinlei; Zhang, Mei; Guo, Min; Wang, Xidong (2008). «Correlation and Prediction of Thermodynamic Properties of Nonaqueous Electrolytes by the Modified TCPC Model». Journal of Chemical & Engineering Data. 53 (1): 149–159. doi:10.1021/je700446q. ISSN 0021-9568.
- ^ Ge, Xinlei; Zhang, Mei; Guo, Min; Wang, Xidong (2008). «Correlation and Prediction of Thermodynamic Properties of Some Complex Aqueous Electrolytes by the Modified Three-Characteristic-Parameter Correlation Model». Journal of Chemical & Engineering Data. 53 (4): 950–958. doi:10.1021/je7006499. ISSN 0021-9568.
- ^ Ge, Xinlei; Wang, Xidong (2009). «A Simple Two-Parameter Correlation Model for Aqueous Electrolyte Solutions across a Wide Range of Temperatures». Journal of Chemical & Engineering Data. 54 (2): 179–186. doi:10.1021/je800483q. ISSN 0021-9568.
- ^ «Project: Ionic Strength Corrections for Stability Constants». IUPAC. Archived from the original on 29 October 2008. Retrieved 2008-11-15.
- ^ Klamt, Andreas (2005). COSMO-RS from quantum chemistry to fluid phase thermodynamics and drug design (1st ed.). Amsterdam: Elsevier. ISBN 978-0-444-51994-8.
- ^ a b N. Butler, James (1998). Ionic equilibrium: solubility and pH calculations. New York, NY [u.a.]: Wiley. ISBN 9780471585268.
- ^ Ellis, A. J.; Golding, R. M. (1963). «The solubility of carbon dioxide above 100 degrees C in water and in sodium chloride solutions». American Journal of Science. 261 (1): 47–60. Bibcode:1963AmJS..261…47E. doi:10.2475/ajs.261.1.47. ISSN 0002-9599.
External links[edit]
- AIOMFAC online-model An interactive group-contribution model for the calculation of activity coefficients in organic–inorganic mixtures.
- Electrochimica Acta Single-ion activity coefficients
Значение, учитывающее термодинамическую неидеальность смесей
Коэффициент активностиявляется фактором используется в термодинамике для учета отклонений от идеального поведения в смеси из химических веществ. В идеальной смеси микроскопические взаимодействия между каждой парой химических частиц одинаковы (или макроскопически эквивалентны, изменение энтальпии раствора и изменение объема при перемешивании равно нулю) и, как результат, свойства смесей могут быть выражены непосредственно через простые концентрации или парциальные давления присутствующих веществ, например Закон Рауля. Отклонения от идеальности компенсируются изменением концентрации на коэффициент активности. Аналогичным образом выражения, включающие газы, могут быть скорректированы на неидеальность путем масштабирования парциальных давлений с помощью коэффициента летучести.
Понятие коэффициента активности тесно связано с понятием активности в химии.
Содержание
- 1 Термодинамическое определение
- 1.1 Ионные растворы
- 2 Экспериментальное определение коэффициентов активности
- 2.1 Радиохимические методы
- 2.2 При бесконечном разбавлении
- 3 Теоретический расчет коэффициентов активности
- 4 Связь с ионным диаметром
- 5 Зависимость от параметров состояния
- 6 Концентрированные растворы электролитов
- 7 Применение к химическое равновесие
- 8 Ссылки
- 9 Внешние ссылки
Термодинамическое определение
химический потенциал, μ B , вещества B в идеальная смесь жидкостей или идеальный раствор определяется как
- μ B = μ B ⊖ + RT ln x B { displaystyle mu _ { mathrm {B}} = mu _ { mathrm {B}} ^ { ominus} + RT ln x _ { mathrm {B}} ,}
где μ. B- химический потенциал чистого вещества B { displaystyle mathrm {B}}и x B — мольная доля th е вещество в смеси.
Это обобщается, чтобы включить неидеальное поведение, записывая
- μ B = μ B ⊖ + RT ln a B { displaystyle mu _ { mathrm {B}} = mu _ { mathrm {B}} ^ { ominus} + RT ln a _ { mathrm {B}} ,}
когда B — это активность вещества в смеси с
- a B = x B γ B { displaystyle a _ { mathrm {B}} = x _ { mathrm {B}} gamma _ { mathrm {B}}}
где γ B — коэффициент активности, который сам может зависеть от x B. Когда γ B приближается к 1, вещество ведет себя так, как если бы оно было идеальным. Например, если γ B ≈ 1, то закон Рауля точен. Для γ B >1 и γ B< 1, substance B shows positive and negative deviation from Raoult’s law, respectively. A positive deviation implies that substance B is more volatile.
Во многих случаях, когда x B стремится к нулю, коэффициент активности вещества B приближается к константе; это соотношение является законом Генри для растворителя. Эти отношения связаны друг с другом посредством уравнения Гиббса – Дюгема. Обратите внимание, что в целом коэффициенты активности безразмерны.
Подробно: Закон Рауля утверждает, что парциальное давление компонента B связано с давлением его пара (давлением насыщения) и его мольной долей x B в жидкости. фаза,
- p B = x B γ B p B σ, { displaystyle p _ { mathrm {B}} = x _ { mathrm {B}} gamma _ { mathrm {B}} p _ { mathrm {B}} ^ { sigma} ;,}
с условием lim x B → 1 γ B = 1. { displaystyle lim _ {x _ { mathrm {B}} to 1} gamma _ { mathrm {B}} = 1 ;.}Другими словами: чистые жидкости представляют собой идеал кейс.
При бесконечном разбавлении коэффициент активности приближается к своему предельному значению, γ B. Сравнение с законом Генри,
- p B = KH, B x B для x B → 0, { displaystyle p _ { mathrm {B}} = K _ { mathrm {H, B}} x _ { mathrm {B}} quad { text {for}} quad x _ { mathrm {B}} to 0 ;,}
немедленно дает
- KH, B = p B σ γ B ∞. { displaystyle K _ { mathrm {H, B}} = p _ { mathrm {B}} ^ { sigma} gamma _ { mathrm {B}} ^ { infty} ;.}
В Другими словами: соединение показывает неидеальное поведение в случае разбавления.
Приведенное выше определение коэффициента активности нецелесообразно, если соединение не существует в виде чистой жидкости. Это часто бывает с электролитами или биохимическими соединениями. В таких случаях используется другое определение, которое рассматривает бесконечное разбавление как идеальное состояние:
- γ B † ≡ γ B / γ B ∞ { displaystyle gamma _ { mathrm {B}} ^ { dagger} Equiv gamma _ { mathrm {B}} / gamma _ { mathrm {B}} ^ { infty}}
с lim x B → 0 γ B † = 1, { displaystyle lim _ {x _ { mathrm {B}} to 0} gamma _ { mathrm {B}} ^ { dagger} = 1 ;,}и
- μ B = μ B ⊖ + RT ln γ B ∞ ⏟ μ B ⊖ † + RT ln (x B γ B †) { displaystyle mu _ { mathrm {B}} = underbrace { mu _ { mathrm {B }} ^ { ominus} + RT ln gamma _ { mathrm {B}} ^ { infty}} _ { mu _ { mathrm {B}} ^ { ominus dagger}} + RT ln left (x _ { mathrm {B}} gamma _ { mathrm {B}} ^ { dagger} right)}
† { displaystyle ^ { dagger}}был использован здесь для различения двух видов коэффициентов активности. Обычно его опускают, так как из контекста ясно, какой вид имеется в виду. Но бывают случаи, когда требуются оба вида коэффициентов активности, которые могут даже входить в одно и то же уравнение, например, для растворов солей в смесях (вода + спирт). Иногда это является источником ошибок.
Изменение мольных долей или концентраций с помощью коэффициентов активности дает эффективные активности компонентов и, следовательно, позволяет применять такие выражения, как закон Рауля и константы равновесия, к обоим идеальные и неидеальные смеси.
Знание коэффициентов активности особенно важно в контексте электрохимии, поскольку поведение растворов электролитов часто далеки от идеального из-за воздействия ионная атмосфера. Кроме того, они особенно важны в контексте химии почвы из-за малых объемов растворителя и, следовательно, высокой концентрации электролитов.
ионных растворов
для растворения Для веществ, которые ионизируют раствор, коэффициенты активности катиона и аниона не могут быть экспериментально определены независимо друг от друга, поскольку свойства раствора зависят от обоих ионов. Коэффициенты активности одного иона должны быть связаны с коэффициентом активности растворенного электролита, как если бы он не диссоциировал. В этом случае используется средний стехиометрический коэффициент активности растворенного электролита γ ±. Он называется стехиометрическим, потому что он выражает как отклонение от идеальности раствора, так и неполную ионную диссоциацию ионного соединения, которая происходит, особенно, с увеличением его концентрации.
Для электролита 1: 1, такого как NaCl, он определяется следующим образом:
- γ ± = γ + γ — { displaystyle gamma _ { pm} = { sqrt { gamma _ {+} gamma _ {-}}}}
где γ + и γ — — коэффициенты активности катиона и аниона соответственно.
В более общем смысле средний коэффициент активности соединения формулы A pBqопределяется как
- γ ± = γ A p γ B qp + q { displaystyle gamma _ { pm} = { sqrt [{p + q}] { gamma _ { mathrm {A}} ^ {p} gamma _ { mathrm {B}} ^ {q}}}}
Коэффициенты одноионной активности можно рассчитать теоретически, например, используя уравнение Дебая – Хюккеля. Теоретическое уравнение можно проверить, комбинируя рассчитанные коэффициенты одноионной активности для получения средних значений, которые можно сравнить с экспериментальными значениями.
Преобладающее мнение о том, что коэффициенты активности одного иона невозможно измерить независимо или, возможно, даже физически бессмысленно, уходит своими корнями в работы Гуггенхайма в конце 1920-х годов. Однако химики никогда не могли отказаться от идеи активности отдельных ионов и, как следствие, коэффициентов активности отдельных ионов. Например, pH определяется как отрицательный логарифм активности ионов водорода. Если преобладающий взгляд на физический смысл и измеримость активности одного иона верен, то определение pH как отрицательного логарифма активности ионов водорода помещает эту величину в категорию неизмеримых. Признавая эту логическую трудность, Международный союз теоретической и прикладной химии (IUPAC) заявляет, что определение pH на основе активности является только условным определением. Несмотря на преобладающее отрицательное мнение об измеримости коэффициентов отдельных ионов, концепция активности отдельных ионов продолжает обсуждаться в литературе, и по крайней мере один автор представляет определение активности одного иона в терминах чисто термодинамических величин и предлагает метод определения активности одного иона. измерение коэффициентов активности одного иона на основе чисто термодинамических процессов.
Экспериментальное определение коэффициентов активности
Коэффициенты активности могут быть определены экспериментально путем проведения измерений на неидеальных смесях. Можно использовать закон Рауля или закон Генри, чтобы получить значение для идеальной смеси, с которым можно сравнить экспериментальное значение для получения коэффициента активности. Также могут использоваться другие коллигативные свойства, такие как осмотическое давление.
Радиохимические методы
Коэффициенты активности могут быть определены радиохимическими методами.
При бесконечном разбавлении
Коэффициенты активности для бинарных смесей равны часто сообщается о бесконечном разбавлении каждого компонента. Поскольку модели коэффициента активности упрощаются при бесконечном разбавлении, такие эмпирические значения могут использоваться для оценки энергий взаимодействия. Примеры приведены для воды:
| X | γx(K) | γW(K) |
|---|---|---|
| Этанол | 4,3800 (283,15) | 3,2800 (298,15) |
| Ацетон | 6,0200 (307,85) |
Теоретический расчет коэффициентов активности
UNIQUAC Регрессия коэффициентов активности (смесь хлороформа / метанола )
Коэффициенты активности растворов электролитов можно рассчитать теоретически, используя уравнение Дебая-Хюккеля или такие расширения, как уравнение Дэвиса, уравнения Питцера или модель TCPC. Также может быть использована теория специфического ионного взаимодействия (SIT).
Для растворов, не содержащих электролитов, можно использовать соответствующие методы, такие как UNIQUAC, NRTL, MOSCED или UNIFAC, при условии, что доступны параметры для конкретного компонента или модели. COSMO-RS — это теоретический метод, который в меньшей степени зависит от параметров модели, поскольку необходимая информация получается из расчетов квантовой механики, специфичных для каждой молекулы (сигма-профили) в сочетании со статистической термодинамической обработкой сегментов поверхности.
Для незаряженных видов коэффициент активности γ 0 в основном соответствует модели высаливания :
- log 10 (γ 0) = b I { displaystyle log _ {10} ( gamma _ {0}) = bI}
Эта простая модель предсказывает активность многих видов (растворенные недиссоциированные газы, такие как CO 2 , H 2 S , NH 3 , недиссоциированные кислоты и основания) до высоких ионной силы (до 5 моль / кг). Значение константы b для CO 2 составляет 0,11 при 10 ° C и 0,20 при 330 ° C.
Для воды в качестве растворителя активность a w можно рассчитать по формуле:
- ln (aw) = — ν b 55,51 φ { displaystyle ln (a _ { mathrm {w}}) = { frac {- nu b} { 55.51}} varphi}
где ν — количество ионов, образующихся в результате диссоциации одной молекулы растворенной соли, b — моляльность соли, растворенной в воде, φ — осмотический коэффициент воды, а константа 55,51 представляет молярность воды. В приведенном выше уравнении активность растворителя (здесь воды) представлена как обратно пропорциональная количеству частиц соли по сравнению с количеством частиц растворителя.
Связь с ионным диаметром
Коэффициент ионной активности связан с ионным диаметром по формуле, полученной из теории Дебая – Хюккеля из электролиты :
- журнал (γ я) = — A zi 2 I 1 + B a I { displaystyle log ( gamma _ {i}) = — { frac {Az_ {i} ^ {2} { sqrt {I}}} {1 + Ba { sqrt {I}}}}}
где A и B — константы, z i — число валентности иона, а I — ионная сила.
Зависимость от параметров состояния
Производная коэффициента активности по температуре связана с избыточной молярной энтальпией соотношением
- H ¯ i E = — RT 2 ∂ ∂ T пер (γ я) { displaystyle { bar {H}} _ {i} ^ { mathsf {E}} = — RT ^ {2} { frac { partial} { partial T}} ln ( gamma _ {i})}
Точно так же производная коэффициента активности по давлению может быть связана с избыточным молярным объемом.
- V ¯ я E знак равно RT ∂ ∂ P пер (γ я) { displaystyle { bar {V}} _ {i} ^ { mathsf {E}} = RT { frac { partial} { partial P}} ln ( gamma _ {i})}
Концентрированные растворы электролитов
Для концентрированных ионных растворов необходимо учитывать гидратацию ионов, как это было сделано Стоксом и Робинсоном в их модель гидратации от 1948 года. Коэффициент активности электролита разделен на электрические и статистические компоненты Э. Глюкауфом, который модифицирует модель Робинсона – Стокса.
Статистическая часть включает индекс гидратации h, количество ионов от диссоциации и отношение r между кажущимся молярным объемом электролита и молярным объемом. воды и моляльности b.
Статистическая часть коэффициента активности концентрированного раствора:
- ln γ s = h — ν ν ln (1 + br 55,5) — h ν ln (1 — br 55,5) + br ( р + час — ν) 55,5 (1 + br 55,5) { displaystyle ln gamma _ {s} = { frac {h- nu} { nu}} ln left (1 + { frac { br} {55.5}} right) — { frac {h} { nu}} ln left (1 — { frac {br} {55.5}} right) + { frac {br (r + h- nu)} {55.5 left (1 + { frac {br} {55.5}} right)}}},
Модель Стокса – Робинсона была проанализирована и улучшена другими исследователями.
Применение к химическому равновесию
В состоянии равновесия сумма химических потенциалов реагентов равна сумме химических потенциалов продуктов. Изменение свободной энергии Гиббса для реакций, Δ r G, равно разнице между этими суммами и, следовательно, в состоянии равновесия равно нулю. Таким образом, для равновесия, такого как
- α A + β B ⇌ σ S + τ T
- Δ r G = σ μ S + τ μ T — (α μ A + β μ B) = 0 { displaystyle Delta _ { mathrm {r}} G = sigma mu _ { mathrm {S}} + tau mu _ { mathrm {T}} — ( alpha mu _ { mathrm {A} } + beta mu _ { mathrm {B}}) = 0 ,}
Подставьте в выражения для химического потенциала каждого реагента:
- Δ r G = σ μ S ⊖ + σ RT ln a S + τ μ T ⊖ + τ RT пер a T — (α μ A ⊖ + α RT пер a A + β μ B ⊖ + β RT пер a B) = 0 { displaystyle Delta _ { mathrm {r}} G = sigma mu _ {S} ^ { ominus} + sigma RT ln a _ { mathrm {S}} + tau mu _ { mathrm {T}} ^ { ominus} + tau RT ln a _ { mathrm {T}} — ( alpha mu _ { mathrm {A}} ^ { ominus} + alpha RT ln a _ { mathrm {A}} + beta mu _ { mathrm {B}} ^ { ominus} + beta RT ln a _ { mathrm {B}}) = 0}
После преобразования это выражение становится
- Δ r G знак равно (σ μ S ⊖ + τ μ T ⊖ — α μ A ⊖ — β μ B ⊖) + RT ln a S σ a T τ a A α a B β = 0 { displaystyle Delta _ { mathrm { r}} G = left ( sigma mu _ { mathrm {S}} ^ { ominus} + tau mu _ { mathrm {T}} ^ { ominus} — alpha mu _ { mathrm {A}} ^ { ominus} — beta mu _ { mathrm {B}} ^ { ominus} справа) + RT ln { frac {a _ { mathrm {S}} ^ { sigma} a _ { mathrm {T}} ^ { tau}} {a _ { mathrm {A}} ^ { alpha } a _ { mathrm {B}} ^ { beta}}} = 0}
Сумма σμ. S+ τμ. T- αμ. A- βμ. B- это стандартное изменение свободной энергии для реакция, Δ r Г. Следовательно,
- Δ r G ⊖ = — RT ln K { displaystyle Delta _ {r} G ^ { ominus} = — RT ln K}
K — константа равновесия. Обратите внимание, что активности и константы равновесия являются безразмерными числами.
Этот вывод служит двум целям. Он показывает взаимосвязь между стандартным изменением свободной энергии и константой равновесия. Это также показывает, что константа равновесия определяется как отношение активности. На практике это неудобно. Когда каждое действие заменяется произведением концентрации и коэффициента активности, константа равновесия определяется как
- K = [S] σ [T] τ [A] α [B] β × γ S σ γ T τ γ A α γ В β { Displaystyle К = { гидроразрыва {[ mathrm {S}] ^ { sigma} [ mathrm {T}] ^ { tau}} {[ mathrm {A}] ^ { alpha} [ mathrm {B}] ^ { beta}}} times { frac { gamma _ { mathrm {S}} ^ { sigma} gamma _ { mathrm {T}} ^ { tau}} { gamma _ { mathrm {A}} ^ { alpha} gamma _ { mathrm {B}} ^ { beta}}}}
где [S] обозначает концентрация S и т. д. На практике константы равновесия определяются в среде, так что коэффициент коэффициента активности постоянен и его можно игнорировать, что приводит к обычному выражению
- K = [S ] σ [T] τ [A] α [B] β { displaystyle K = { frac {[ mathrm {S}] ^ { sigma} [ mathrm {T}] ^ { tau}} {[ mathrm {A}] ^ { alpha} [ mathrm {B}] ^ { beta}}}}
, который применяется при условиях, когда коэффициент активности имеет определенное (постоянное) значение.
Ссылки
Внешние ссылки
- Онлайн-модель AIOMFAC Интерактивная модель группового вклада для расчета коэффициентов активности в органических-неорганических смесях.
- Electrochimica Acta Коэффициенты одноионной активности
Как найти средний ионный коэффициент активности?
и средний ионный коэффициент активности γ±: lgγ± = -A⏐z+ z−⏐ I , (10) Page 5 5 где z+ и z− — заряды катиона и аниона, I — ионная сила раствора, А — константа, зависящая от диэлектрической проницаемости растворителя и температуры.
Как найти активность электролита?
Активность иона ai выражается в виде произведения концентрации иона mi на его коэффициент активности i: ai = i mi. где n = n + + n -.
Что такое ионная сила раствора?
Ионная сила раствора (I, размерность концентрации) – это полусумма произведений концентраций всех ионов в растворе на квадрат их заряда.
Что такое активность электролита?
Переносчиками электрического тока в растворах электролитов являются ионы, образующиеся при диссоциации молекул электролитов. Поскольку при диссоциации число частиц в растворе возрастает, растворы электролитов обладают аномальными коллигативными свойствами.
Как определить коэффициент активности?
сохранялась и для реальных растворов: GA = GA0 + nRT·ln aA. Следовательно, активность — это та концентрация, которую имел бы компонент воображаемого идеального раствора, обладающего теми же термодинамическими свойствами, что и данный реальный раствор и имеет размерность (моль/л).
В чем измеряется активность ионов?
Численно эта величина выражается как концентрация С(Н+) в одном литре изучаемого раствора и измеряется в моль/литр или граммах/литр. Абсолютно чистую воду принято считать нейтральной, так как концентрация ионов Н+ и ОН– в ней равна.
Как определить активность вещества?
Методы определения активности
- По равновесному давлению пара
- По повышению температуры кипения раствора
- По понижению температуры замерзания раствора
- По осмотическому давлению раствора
- По распределению компонента между конденсированными фазами
- По равновесию химической реакции с газовой фазой
Что такое солевой эффект?
солевой эффект — – влияние ионной силы раствора на константу скорости реакций между ионами (первичный солевой эффект) и на равновесные концентрации ионов (вторичный солевой эффект).
Что такое ионная атмосфера?
повышенная концентрация ионов противоположного знака в объёме, окружающем данный ион в растворе; образуется вследствие действия электрического поля, создаваемого этим ионом.
Что называется активностью?
Акти́вность компонентов раствора — эффективная (кажущаяся) концентрация компонентов с учётом различных взаимодействий между ними в растворе, то есть с учётом отклонения поведения системы от модели идеального раствора.
Как определить ионную силу раствора?
Например, для раствора NaCl с концентрацией 0,001 моль/л, в котором присутствуют два вида однозарядных ионов Na+ и Cl− с концентрациями также равными 0,001 моль/л, ионная сила будет вычисляться следующим образом: I(NaCl) = 0,5(z²(Na+)•c(Na+) + z²(Cl−)•c(Cl−)) = 0,5(1²•c(NaCl) + (-1)²•c(NaCl)) = c(NaCl)
В чем измеряется коэффициент активности?
сохранялась и для реальных растворов: GA = GA0 + nRT·ln aA. Следовательно, активность — это та концентрация, которую имел бы компонент воображаемого идеального раствора, обладающего теми же термодинамическими свойствами, что и данный реальный раствор и имеет размерность (моль/л).
В чем измеряется активность?
беккерель
Единицы измерения активности В Международной системе единиц (СИ) единицей активности является беккерель (русское обозначение: Бк; международное: Bq); 1 Бк = с−1. В образце с активностью 1 Бк происходит в среднем 1 распад в секунду.
Как найти активность препарата?
- число радиоактивных ядер уменьшается в е = 2,71.
- раз: τ = 1/ λ
- Период полураспада T1/2 – время, за которое число
- радиоактивных ядер уменьшается вдвое.
- Активность препарата = Число распадов в единицу
- времени I = dN / dt = N λ
Сколько лет идет распад радиации?
Период полураспада полония 214 составляет одну секунду, в то время как урана 238 – 4,5 миллиарда лет. Кривая радиоактивного распада: через два периода радиоактивность вещества снижается в четверо, через три – в восемь раз и т. д.
Какое из следующих положений относится к теории сильных электролитов Дебая Гюккеля )?
(теория Дебая—Хюккеля) Основные положения: 1) Электролит всегда полностью диссоциирован (α = 1). 2) Растворитель – это континуум с диэлектрической проницаемостью ε. 3) Ионы участвуют ТОЛЬКО в 2 процессах: хаотическом тепловом движении и электростатическом (кулоновском) взаимодействии друг с другом.
Что называется активности радиоактивного изотопа?
Акти́вность радиоакти́вного исто́чника — число элементарных радиоактивных распадов в единицу времени.
В химической термодинамике, активность (символ a) является мерой » эффективная концентрация разновидностей в смеси, в том смысле, что химический потенциал разновидностей зависит от активности реального раствора точно так же, как это будет зависеть от концентрации для идеальное решение. Термин «активность» в этом смысле был введен американским химиком Гилбертом Н. Льюисом в 1907 году.
По соглашению, активность рассматривается как безразмерная величина, хотя его значение зависит от обычного выбора стандартного состояния для вида. Активность чистых веществ в конденсированных фазах (твердых или жидких) обычно принимается за единица (цифра 1). Активность зависит, помимо прочего, от температуры, давления и состава смеси. Для газов активность представляет собой эффективное парциальное давление и обычно обозначается как летучесть.
. Разница между активностью и другими показателями состава возникает из-за того, что молекулы в неидеальных газах или решения взаимодействуют друг с другом, притягивая или отталкивая друг друга. На активность иона иона особенно влияет его окружение.
Действия следует использовать для определения констант равновесия, но на практике вместо них часто используются концентрации. То же самое часто относится к уравнениям для скорости реакции. Однако бывают обстоятельства, при которых активность и концентрация существенно различаются, и, как таковые, нельзя приближать к концентрациям там, где требуется активность. Два примера служат, чтобы проиллюстрировать этот момент:
- В растворе иодата калия KH (IO 3)2при 0,02 M активность на 40% ниже, чем рассчитанный ион водорода pH, чем ожидалось.
- Когда 0,1 M раствор соляной кислоты, содержащий метиловый зеленый индикатор добавляется к 5 М раствору хлорида магния, цвет индикатора меняется с зеленого на желтый, что указывает на повышение кислотности, хотя на самом деле кислота была разбавлена. Хотя при низкой ионной силе (< 0.1 M) the коэффициент активности приближается к единице, этот коэффициент может фактически увеличиваться с увеличением ионной силы в режиме высокой ионной силы. Для растворов соляной кислоты минимум составляет около 0,4 М.
Содержание
- 1 Определение
- 1,1 Коэффициент активности
- 2 Стандартные состояния
- 2.1 Газы
- 2.2 Смеси в целом
- 2.3 Разбавленные растворы (неионные)
- 2.4 Ионные растворы
- 3 Измерение
- 3.1 Действие одного иона Еще раз об измеримости активности
- 4 Использование
- 5 Примеры значений
- 6 См. также
- 7 Ссылки
- 8 Внешние ссылки
Определение
Относительная активность вида i, обозначенная a i, определяется как:
- ai = e μ i — μ i ⊖ RT { displaystyle a_ {i} = e ^ { frac { mu _ {i} — mu _ {i} ^ { ominus}} {RT}}}

где μ i — (молярный) химический потенциал вида i в интересующих условиях, μ. i- (молярный) химический потенциал этого вещества при некотором определенном наборе стандартных условий, R — газовая постоянная, T — термодинамическая температура, а e — экспоненциальная константа.
В качестве альтернативы это уравнение можно записать как:
- μ i = μ i ⊖ + RT ln ai { displaystyle mu _ {i} = mu _ {i} ^ { ominus} + RT ln {a_ {i}}}

В общем, активность зависит от любого фактора, который изменяет химический потенциал. К таким факторам могут относиться: концентрация, температура, давление, взаимодействия между химическими веществами, электрические поля и т. Д. В зависимости от обстоятельств некоторые из этих факторов могут быть более важными, чем другие.
Действие зависит от выбора стандартного состояния, так что изменение стандартного состояния также изменит действие. Это означает, что активность — это относительный термин, который описывает, насколько «активное» соединение по сравнению с тем, когда оно находится в стандартных условиях состояния. В принципе, выбор стандартного состояния произвольный; однако его часто выбирают из соображений математического или экспериментального удобства. В качестве альтернативы также можно определить «абсолютную активность», λ, которая записывается как:
- λ i = e μ i RT { displaystyle lambda _ {i} = e ^ { frac { mu _ {i}} {RT}} ,}

Коэффициент активности
Коэффициент активности γ, который также является безразмерной величиной, связывает активность с измеренной долей количества xi(или y i в газовой фазе), моляльность bi, массовая доля wi, количественная концентрация ciили массовая концентрация ρi:
- ai = γ x, ixi = γ b, ibib ⊖ знак равно γ вес, iwi знак равно γ с, icic ⊖ знак равно γ ρ, я ρ я ρ ⊖ { displaystyle a_ {i} = gamma _ {x, i} x_ {i} = gamma _ {b, i} { frac {b_ {i}} {b ^ { ominus}}} , = gamma _ {w, i} w_ {i} = gamma _ {c, i} { frac {c_ {i}} {c ^ { ominus}}} , = gamma _ { rho, i} { frac { rho _ {i}} { rho ^ { ominus}}} ,}

Деление на стандартную молярность b или стандартную количественную концентрацию c необходимо, чтобы гарантировать, что и активность, и коэффициент активности безразмерны, как это принято.
Активность одинакова независимо от способы выражения композиции и стандартное состояние выбраны таким образом, чтобы приведенные выше выражения были равны.
Когда коэффициент активности близок к 1, вещество показывает почти идеальное поведение в соответствии с законом Генри. В этих случаях активность может быть заменена соответствующей безразмерной мерой состава x i, b i / b или c i / c. Также возможно определить коэффициент активности в терминах закона Рауля : Международный союз теоретической и прикладной химии (IUPAC) рекомендует символ f для этого коэффициента активности, хотя это должно не следует путать с летучестью.
- ai = fixi { displaystyle a_ {i} = f_ {i} x_ {i} ,}

Стандартные состояния
Газы
В большинстве лабораторных ситуаций разница в поведении реального и идеального газа зависит только от давления и температуры, а не от наличия каких-либо других газов. При данной температуре «эффективное» давление газа i определяется его летучестью fi: оно может быть выше или ниже его механического давления. Исторически сложилось так, что у бегунов есть измерение давления, поэтому безразмерная активность определяется как:
- ai = fip ⊖ = φ iyipp ⊖ { displaystyle a_ {i} = { frac {f_ {i}} {p ^ { ominus}}} = varphi _ {i} y_ {i} { frac {p} {p ^ { ominus}}}}

где φ i — безразмерный коэффициент летучести вида y i — его доля в газовой смеси (y = 1 для чистого газа), а p — полное давление. Значение p является стандартным давлением: оно может быть равно 1 атм (101,325 кПа) или 1 бар (100 кПа) в зависимости от источника данных., и его всегда следует указывать.
Смеси в целом
Наиболее удобный способ выразить состав общей смеси — использовать количественные доли x (обозначаемые y в газовой фазе) различных компонентов, где
- xi = nin, ∑ ixi = 1 { displaystyle x_ {i} = { frac {n_ {i}} {n}} ,, qquad sum _ {i} x_ {i} = 1 ,}

Стандартным состоянием каждого компонента в смеси считается чистое вещество, т.е. чистое вещество имеет активность, равную единице. Когда используются коэффициенты активности, они обычно определяются в терминах закона Рауля,
- ai = fixi { displaystyle a_ {i} = f_ {i} x_ {i} ,}
где f i — коэффициент активности по закону Рауля: коэффициент активности, равный единице, указывает на идеальное поведение в соответствии с законом Рауля.
Разбавленные растворы (неионогенные)
Растворенное вещество в разбавленном растворе обычно следует закону Генри, а не закону Рауля, и обычно выражают состав раствора с точки зрения количественной концентрации c (в моль / л) или моляльности b (в моль / кг) растворенного вещества, а не количественных долей. Стандартным состоянием разбавленного раствора является гипотетический раствор с концентрацией c = 1 моль / л (или молярностью b = 1 моль / кг), который демонстрирует идеальное поведение (также называемое поведением «бесконечного разбавления»). Стандартное состояние и, следовательно, активность зависят от того, какая мера состава используется. Моляльность часто является предпочтительной, поскольку объемы неидеальных смесей не являются строго аддитивными и также зависят от температуры: моляльность не зависит от объема, в то время как количественные концентрации зависят.
Активность растворенного вещества определяется по формуле:
- ac, i = γ c, icic ⊖ ab, i = γ b, ibib ⊖ { displaystyle { begin {align} a_ {c, i} = gamma _ {c, i} , { frac {c_ {i}} {c ^ { ominus}}} \ [6px] a_ {b, i} = gamma _ {b, i} , { frac {b_ {i}} {b ^ { ominus}}} end {align}}}
![{ displaystyle { begin {align} a_ {c, i} = gamma _ {c, i} , { fr ac {c_ {i}} {c ^ { ominus}}} \ [6px] a_ {b, i} = gamma _ {b, i} , { frac {b_ {i}} {b ^ { ominus}}} end {align}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/fc8d9ae34964a1970506f129464ec7d6f631d1e4)
Ионные растворы
Когда растворенное вещество подвергается ионной диссоциации в растворе (например, в соли), система становится явно неидеальной, и нам нужно принять во внимание процесс диссоциации. Можно определить активности для катионов и анионов отдельно (a + и -).
В жидком растворе коэффициент активности данного иона (например, Са) невозможно измерить, поскольку экспериментально невозможно независимо измерить электрохимический потенциал иона в растворе. (Нельзя добавлять катионы, не добавляя одновременно анионы). Поэтому вводится понятие
- средней ионной активности
- a. ±= a. +a. −
- средней ионной молярности
- b. ±= b. +b. −
- среднего коэффициента ионной активности
- γ. ±= γ. +γ. −
, где ν = ν + + ν — представляют стехиометрические коэффициенты, участвующие в процессе ионной диссоциации
, хотя γ + и γ — не могут определяется отдельно, γ ± — это измеримая величина, которая также может быть предсказана для достаточно разбавленных систем с помощью теории Дебая – Хюккеля. Для растворов электролитов с более высокими концентрациями теорию Дебая-Хюккеля необходимо расширить и заменить, например, моделью раствора электролита Питцера (см. внешние ссылки ниже для примеров). Для активности сильного ионного растворенного вещества (полная диссоциация) мы можем записать:
- a2= a. ±= γ. ±m. ±
Измерение
Самый прямой способ измерения активности летучих частиц — это измерить его равновесное парциальное давление пара. Для нелетучих компонентов, таких как сахароза или хлорид натрия, этот подход не работает, поскольку они не имеют измеряемого давления пара при большинстве температур. Однако в таких случаях вместо этого можно измерить давление паров растворителя. Используя соотношение Гиббса – Дюгема, можно преобразовать изменение давления паров растворителя с концентрацией в активность растворенного вещества.
Самый простой способ определить, как активность компонента зависит от давления, — это измерить плотность раствора, зная, что реальные растворы имеют отклонения от аддитивности (молярных) объемов чистых компонентов по сравнению с (молярными) объем раствора. Это включает использование парциальных молярных объемов, которые измеряют изменение химического потенциала по отношению к давлению.
Другой способ определения активности вида — это манипулирование коллигативными свойствами, в частности, понижением точки замерзания. Используя методы снижения точки замерзания, можно вычислить активность слабой кислоты по соотношению
- b ‘= b (1 + a) { displaystyle b ^ { prime} = b (1 + a) ,}

где b ′ — общая равновесная моляльность растворенного вещества, определенная любым измерением коллигативных свойств (в данном случае ΔT fus, b — номинальная моляльность, полученная при титровании, а a — активность вещества..
Существуют также электрохимические методы, позволяющие определять активность и ее коэффициент.
Значение среднего коэффициента ионной активности γ ± ионов в растворе также можно оценить с помощью уравнения Дебая – Хюккеля, уравнения Дэвиса или уравнения Питцера.
Повторное рассмотрение измеримости активности одного иона
Преобладающее мнение о том, что активность отдельных ионов неизмеримо или, возможно, даже физически бессмысленно, уходит корнями в работы Гуггенхайма в конце 1920-х годов. Однако химики так и не смогли определить u p идея активности одного иона. Например, pH определяется как отрицательный логарифм активности ионов водорода. Подразумевается, что если преобладающий взгляд на физический смысл и измеримость активности одного иона верен, он относит pH к категории термодинамически неизмеримых величин. По этой причине Международный союз теоретической и прикладной химии (IUPAC) утверждает, что определение pH на основе активности является только условным определением, и далее заявляет, что установление первичных стандартов pH требует применения концепции «первичного метода измерения», привязанного к ячейке Харнеда. Тем не менее, концепция активности отдельных ионов продолжает обсуждаться в литературе, и по крайней мере один автор претендует на определение активности отдельных ионов в терминах чисто термодинамических величин. Тот же автор также предлагает метод измерения коэффициентов активности отдельных ионов, основанный на чисто термодинамических процессах.
Использование
Химическая активность должна использоваться для определения химических потенциалов, где химический потенциал зависит от температуры T, давления p и активности a i согласно формуле :
- μ i = μ i ⊖ + RT ln ai { displaystyle mu _ {i} = mu _ {i} ^ { ominus} + RT ln {a_ {i}}}
где R — газовая постоянная и μ. i- значение μ i при стандартных условиях. Обратите внимание, что выбор шкалы концентрации влияет как на активность, так и на химический потенциал стандартного состояния, что особенно важно, когда исходным состоянием является бесконечное разбавление растворенного вещества в растворителе.
Формулы, включающие активности, можно упростить, если учесть, что:
- Для химического раствора:
- растворитель имеет активность, равную единице (только верное приближение для разбавленные растворы)
- При низкой концентрации активность растворенного вещества может быть приблизительно выражена отношением его концентрации к стандартной концентрации:
-
-
- ai = cic ⊖ { displaystyle a_ {i} = { frac {c_ {i}} {c ^ { ominus}}}}
- ai = cic ⊖ { displaystyle a_ {i} = { frac {c_ {i}} {c ^ { ominus}}}}
-

Следовательно, он примерно равен его концентрации.
- Для смеси газа при низком давлении активность равна отношению парциального давления газа к стандартному давлению:
-
- ai = pip ⊖ { displaystyle a_ {i} = { frac {p_ {i}} {p ^ { ominus}}}}
- ai = pip ⊖ { displaystyle a_ {i} = { frac {p_ {i}} {p ^ { ominus}}}}
- Следовательно, оно равно парциальному давлению в атмосферах (или барах) по сравнению со стандартным давление в 1 атмосферу (или 1 бар).

- Для твердого тела однородное твердое тело одного вида при давлении 1 бар имеет активность, равную единице. То же самое верно и для чистой жидкости.
Последнее следует из любого определения, основанного на законе Рауля, потому что, если мы позволим концентрации растворенного вещества x 1 стать равным нулю, давление пара растворителя p будет перейти к p *. Таким образом, его активность a = p / p * упадет в единицу. Это означает, что если во время реакции в разбавленном растворе образуется больше растворителя (например, при реакции образуется вода), мы обычно можем установить его активность равной единице.
Активность твердой и жидкой фаз не очень сильно зависит от давления, поскольку их молярные объемы обычно малы. Графит при 100 барах имеет активность только 1,01, если мы выберем p = 1 бар в качестве стандартного состояния. Только при очень высоких давлениях можно беспокоиться о таких изменениях.
Примеры значений
Примерные значения коэффициентов активности хлорида натрия в водном растворе приведены в таблице. В идеальном решении все эти значения равнялись бы единице. Отклонения имеют тенденцию увеличиваться с увеличением моляльности и температуры, но с некоторыми исключениями.
-
Моляльность (моль / кг) 25 ° C 50 ° C 100 ° C 200 ° C 300 ° C 350 ° C 0,05 0,820 0,814 0,794 0,725 0,592 0,473 0,50 0,680 0,675 0,644 0,619 0,322 0,182 2,00 0,669 0,675 0,641 0,450 0,212 0,074 5,00 0,873 0,886 0,803 0,466 0,167 0,044
См. Также
 Химический портал
Химический портал
- Летучесть, эквивалент активности для парциального давления
- Химическое равновесие
- Электрохимический потенциал
- Избыточный химический потенциал
- Частичное молярное свойство
- Термодинамическое равновесие
- Тепловое расширение
- Вириальное расширение
- Активность воды
- Неслучайная двухжидкостная модель (модель NRTL) — расчеты фазового равновесия
- Модель UNIQUAC — расчет фазового равновесия
Ссылки
Ext ссылки
- Эквивалентность различных форм коэффициентов активности и химических потенциалов
- Расчет коэффициентов активности обычных неорганических электролитов и их смесей
- Онлайн-модель AIOMFAC : калькулятор коэффициентов активности неорганических ионов, воды и органические соединения в водных растворах и многокомпонентные смеси с органическими соединениями.