Физика
Выполнил студент
группы 2870
Лабораторная
работа № 4
Определение энеогии диссоциации молекул по полосатому спектру поглощения
Цель работы:
изучение спектра двухатомных
молекул йода ил брома в видимой области
части спектра; определение спектральным
методом энергии диссоциации молекул.
Основные положения теории двухатомных молекул
Изучение спектров
молекул и получение данных о свойствах
молекул составляет содержание раздела
физики – молекулярной спектроскопии.
Строение молекулы
сложнее строения атомов, из которых она
образована. Это отражается на структуре
ее энергетических уровней и на спектре
поглощения и излучения молекул. Объяснение
характера молекулярных спектров дает
квантовая механика.
Полная энергия
молекулы W
в каком либо стационарном состоянии
складывается из электронной энергии
We
, обусловленной электронной конфигурацией
молекулы, колебательной Wυ
и вращательной Wy
энергии, зависящих от колебаний и
вращения атомов относительно их общего
центра инерции.
W=We + W υ +Wy
Все три вида энергии
квантуются, т.е. могут принимать лишь
определенные дискретные значения.
Значение энергии
We
близки к тем значениям энергии, которыми
могут обладать электроны изолированных
атомов, образующих молекулу.
Энергия колебания
ядер в молекуле характеризуется
выражением:
где υ
— колебательное квантовое число,
принимающее значение 0,1,2,3,… ;
— частота
собственных колебаний молекулы; χe
— постоянная ангармоничности, учитывающая,
что при интенсивных колебаниях
( больших υ)
молекулу нельзя рассматривать как
гармонический осциллятор; h
– постоянная Планка.
Энергия вращательного
движения молекулы определяется формулой:

где — вращательное
квантовое число, принимающее значение
0,1,2,3…;
—
момент инерции молекулы; М – приведенная
масса, равная M
=
![]()
; М![]()
и М![]()
— массы ядер; ρ![]()
— межъядерное расстояние.
Энергия кванта
испускаемого или поглощаемого молекулой,
равна:
где ΔWe
, ΔW
υ
, ΔWy
— изменения соответствующих частей
энергии молекулы. Опыт и теория показывают,
что
В невозбужденном
состоянии молекулы все три вида энергии
имеют минимальное значение. При слабых
возбуждениях изменяется только
вращательная энергия Wy,
при более сильных возбуждениях изменяется
W
при изменении
электронной конфигурации молекулы
изменяется и соответствующая энергия
We.
При таких сильных воздействиях
одновременно может измениться
колебательная и вращательная энергия
молекулы. На рис. 1 показана система
колебательных и вращательных уровней
для двух электронных состояний. При
переходе молекулы из одного электронного
состояния в другое каждое колебательное
состояние верхнего электронного
состояния может комбинироваться с
каждым колебательным состоянием нижнего
электронного состояния.
Строгого правила
отбора для колебательных квантовых
чисел не существует. Такие переходы
называются электронно-колебательными.
При не высоких
температурах (Т~300К) большая часть
молекулы находится на нижнем колебательном
уровне (υ
= 0) основного электронного состояния.
При поглощении соответствующих квантов
энергии практически наблюдается только
переходы из нижнего колебательного
состояния (υ
= 0) на различные колебательные уровни
(υ)
верхнего электронного состояния.
Если величина
колебательного кванта в основном
состоянии не велика или температура
достаточно велика, то есть часть молекул
находится в возбужденном состоянии (υ
= 1).
Могут возникнуть
электронно-колебательные переходы из
колебательного состояния с υ
= 1 на различные колебательные уровни
верхнего электронного состояния υ
.
Для получения
спектра поглощения молекул исследуемое
вещество освещают от источника, спектр
излучения которого сплошной.
Схема рис.2,а
поясняет возникновение
электронно-колебательного полосатого
спектра поглощения двухатомных молекул.
Н а
а
рисунке изображены энергетические
уровни двух электронных состояний
молекулы We
и We
. Изменение энергии, соответствующее
этим состояниям, — ΔWe.
Для каждого электронного уровня показано
несколько колебательных уровней,
характеризуемых квантовыми числами
υ
и υ
соответственно.
Вращательные
уровни на схеме не показаны. Если бы
энергия вращения молекул не изменялась,
то спектр поглощения молекул состоял
бы из системы темных линий поглощения
на светлом фоне сплошного спектра
излучения источника света.
Каждому колебательному
уровню соответствует система вращательных
уровней энергии, поэтому полосы
электронно-колебательного спектра
состоят из близко расположенных линий
поглощения. Квантовые переходы,
затрагивающие изменение колебательной
и вращательной энергий, называют
колебательно-вращательными переходами.
Соответствующие им спектры —
колебательно-вращательные.
Е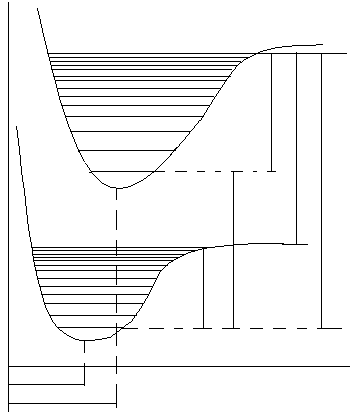 сли
сли
интенсивность монотонно изменяется в
пределах полосы от наименьшего до
наибольшего, то отчетливо видна граница
между соседними полосами. Ее называют
кантом электронно-колебательной полосы.
П риборы
риборы
средней разрешающей способности
вращательную структуру не разрешают.
На рис. 2,б представлена структура
полосатого спектра поглощения двухатомных
молекул, который можно получить с помощью
таких спектральных приборов.
К электронно-колебательному
полосатому спектру в коротковолновой
области спектра примыкает сплошной
спектр поглощения. Обозначим
частоту излучения,
при поглощении которого полосатый
спектр поглощения переходит в сплошной.
Граничная частота
в спектре поглощения соответствует
энергии поглощения фотона ,
достаточной для диссоциации молекулы.
Для понимания
спектрального метода определения
энергии диссоциации молекул по полосатому
электронно-колебательному спектру
поглощения рассмотрим кривые потенциальной
энергии молекул.
Кривая потенциальной
энергии двухатомной невозбужденной
молекулы U(r)
показана на рис.3. На оси абсцисс
указано расстояние между ядрами атомов
молекул r,
на оси ординат – соответствующая
энергия. При r
= r![]()
состояние молекулы наиболее
устойчиво, так как сила притяжения,
возникающая благодаря электронной
связи атомов, уравновешивается кулоновским
отталкиванием одновременно заряженных
ядер. Потенциальная энергия минимальна.
Если возбуждены
колебательные состояния молекул, то
ядра атомов в молекуле совершают
колебания относительно положения
равновесия r![]()
.
Разрешенные
значения колебательной энергии
представлены на рис.3 горизонтальными
линиями. Амплитуды колебаний определяются
точками пересечения кривой потенциальной
энергии уровнями колебательной энергии.
Кривая потенциальной энергии молекулы
при малых значениях υ
может быть
представлена кривой потенциальной
энергии гармонического осциллятора
(пунктирная линия).
При увеличении
колебательной энергии молекулы отклонение
ядер от положения равновесия увеличивается.
Кривая потенциальной энергии асимптотически
приближается к предельному значению
U(∞)
при r →
.
Межатомные связи
ослабевают, происходит диссоциация
молекул. Энергии диссоциации соответствует
наибольшая энергия колебаний.
Большие порции
энергии, достаточные для диссоциации
молекул, могут вызвать возбуждение ее
электронной оболочки. Возбуждение
электронной оболочки резко меняет вид
силового поля, а следовательно, вид
кривой потенциальной энергии U(r)
(рис.3). В возбужденном состоянии прочность
связи меньше, равновесное расстояние
r![]()
между ядрами больше, чем для основного
состояния. При увеличении колебательной
энергии увеличивается расстояние между
ядрами. При r →
кривая потенциальной энергии стремиться
к пределу ,соответствующему
энергии диссоциации возбужденной
молекулы D
<
D
(рис.3).
Наименьшую энергию кванта ,
поглощенного невозбужденной молекулой,
достаточную для изменения электронного
состояния молекулы ΔWe
и диссоциации возбужденной молекулы,
можно представить равенством:
Энергия возбуждения
молекулы
ΔWe
переходит
в энергию возбуждения электронных
оболочек и кинетическую энергию
разлетающихся атомов. Диссоциация
сопровождается свечением атомов.
Из формулы известно,
что молекулы голоиодов, находящихся в
возбужденном электронном состоянии,
диссоциируют на один возбужденный и
один невозбужденный атом с энергией W
. Поэтому процесс диссоциации в
возбужденном состоянии можно рассматривать
как диссоциацию молекулы в основном
состоянии и возбуждение одного из атомов
до энергии W
. В этом случае можно написать :
Энергия диссоциации
молекулы
D может
быть найдена, если известна энергия W
и экспериментально определена граничная
частота полосатого спектра
(см.рис.3):
Если энергия
поглощения фотона , то
избыток энергии при диссоциации идет
на увеличение кинетической энергии
возбужденных атомов. Кинетические
энергии атомов могут принимать любые
значения, поэтому спектр поглощения
молекул при — непрерывный.
Расчет энергии связи кристалла выполняется таким же путем, как и вычисление энергии диссоциации молекулы. Выразим параметр В через равновесное расстояние го между [c.24]
Пример 63.1. Найти энергию диссоциации молекулы дейтерия Dj, если энергия диссоциации молекулы водорода Hj равна 4,48 эВ, а энергия основного колебательного уровня составляет 0,26 iB. [c.324]
Энергию уровня в потенциальной яме, при которой происходит диссоциация молекулы Н2, будем считать равной нулю. Ясно, что на этом уровне осуществляется также и диссоциация молекулы дейтерия. Дно потенциальной ямы ниже этого уровня на (0,48 + 0,26) эВ. На такой же глубине находится и дно потенциальной ямы молекулы дейтерия. Первый колебательный уровень молекулы дейтерия имеет энергию 0,26(Шр/то) = 0,18 эВ. Энергия диссоциации молекулы дейтерия (4,48 + 0,26 — 0,18) эВ = 4,56 эВ. [c.324]
По определенным таким образом энергиям диссоциации одних молекул можно рассчитать энергии диссоциации других молекул, образующихся из них в результате химических реакций, если известны тепловые эффекты этих реакций. Так, по определенным из спектроскопических данных энергиям диссоциации О2 и N2 и термохимической теплоте образования N0 можно рассчитывать энергию диссоциации молекулы N0. [c.66]
Известно, что энтальпия торможения продуктов сгорания высока и распределена по различным степеням свободы молекул в виде поступательного, вращательного и колебательного движений, а также энергии диссоциации молекул (энергию электронного возбуждения и ионизации можно не рассматривать, так как температуры в камере сгорания не настолько высоки). [c.20]
Энергия диссоциации молекул и ионизации атомов различных газов [c.237]
Элемент Энергия диссоциации молекулы, эВ Энергия ионизации атома, эВ [c.237]
Энергии диссоциации молекул…..425 [c.4]
ЭНЕРГИИ ДИССОЦИАЦИИ МОЛЕКУЛ [c.425]
Подобным образом можно определить также энергию диссоциации молекулы в возбужденном состоянии [c.85]
Рис. 1.38. а — Определение энергии диссоциации молекулы методом графической экстраполяции (заштрихованная площадь дает нижний предел Оо) б и в — фрагменты графиков, на которых значения АО отсчитываются от У + 7г и [c.85]
Энергия диссоциации димера на две молекулы уксусной кислоты (разрываются две связи) составляет 58 кДж/моль, что близко к общепринятой энергии слабой химической связи молекул, не содержащих водород. Например, энергия диссоциации молекулы Ыа2 = 71, КЬ2=41, ХеО = 34, ХеМ = 23, Mg2=4,8 кДж/моль. [c.214]
Определите энергию диссоциации молекулы Ь н методом графической экстраполяции и методом линейной экстраполяции, если сое=378,72 см-, соеД е = 2,98 см . Результаты сравните. Нарисуйте систему переходов между электронно-колебательными состояниями. [c.239]
Следует отметить, что в настоящем исследовании излучательной способности цезиевой плазмы не рассматривался процесс молекулярного излучения, что прежде всего связано с отсутствием необходимых спектроскопических данных (сил осцилляторов, множителей Франка—Кондона и т. д.). Однако ввиду малой энергии диссоциации молекулы s2 (- 0,45 эв [14]) можно предположить, что вклад этого процесса излучения при малых давлениях ата) пре- [c.308]
Воспользоваться значением энергии диссоциации молекулы водорода 2е — е = 4,47 эв. [c.165]
Энергия диссоциации. По масс-спектрометрическим данным, энергия диссоциации молекулы РЬТе о = 51 400 2000 кал/моль [181]. В справочнике [50] рекомендуется Од = = 54 702 1900 кал/моль. [c.138]
Энергия диссоциации. В настоящее время для энергии диссоциации молекулы В Те рекомендуется [50] [c.250]
Азот в зону сварки проникает из воздущной атмосферы, где он находится в молекулярном состоянии. Энергия диссоциации молекулы азота очень велика и составляет [c.337]
Последнее выражение позволяет вычислить разность энергий между стационарным равновесным состоянием молекулы (ЫаС1) и состоянием, в котором два иона разделены бесконечно большим расстоянием. Эта разность энергий называется энергией диссоциации молекулы (О). Минимальное значение потенциальной энергии для кристалла будет [c.23]
Равновесное расстоянне. Расстояние, при котором достигает минимума, есть равновесное расстояние между атомами в молекуле водорода, а соответствующая энергия является энергией диссоциации молекулы водорода. Из эксперимента.льных данных следует, что равновесное расстояние между атомами в молекуле водорода равно 1,4 йо, а энергия диссоциации равна 4,5 эВ. Теоретические расчеты дают удовлетворительное согласие с этими величинами. Наличие сил отталкивания между атомами с параллельными спинами также было обнаружено экспериментально. В частности, при столкновении атомы могут образовывать молекулу лишь тогда, когда спины электронов анти-параллельны. Следовательно, при столкновении двух атомов вероятность того, что между ними будут действовать силы притяжения, равна V4, в то время как вероятность возникновения сил отталкивания равна /4. Это обусловлено тем, что имеются три спиновые волновые функции для триплетного состояния и [c.311]
Аюо) = с ав представляет энергию диссоциации молекулы АВ на атомы. Разность А(Аюо) — А(Маоо = = представляет собой количество теплоты, возникающее при хемосорбции одной молекулы. [c.82]
В обычных условиях А.— двухатомный газ. хМоле-кула N2 диамагнитна. Площадь, занимаемая ею при адсорбции на поверхности твердых тел, принята равной 0,162 нм . Энергия диссоциации молекулы велика и составляет при О К 941,6 0,й кДж/ыоль. [c.32]
Определите энергию диссоциации молекулы Нг методом экстраполяции Берджа — Шпонер и рассчитайте энергию диссоциации ее изотопа 02, пренебрегая второй ангармоничностью. [c.240]
Энергии диссоциации молекул 7 равны нескольким электроновольтам (у кислорода 7 = 5,11 е, у азота 9,74 у окиси азота 6,5 Молекулы диссоциируют обычно при температурах выше 3000°, важных с практической точки зрения. Температуры в несколько тысяч градусов получаются при движении тел в атмосфере с гиперзвуковыми скоростями. Неравновесная диссоциация в воздухе (так же как и колебательная релаксация) существенным образом влияет на обтекание тела и теплообмен, и потому кинетика диссоциации, в особенности диссоциации компонентов воздуха, изучалась многими авторами. [c.228]
Наводороживание металла. Разрушающее действие водорода на сталь заслуживает швышевного инимания, особенно Б связи с разрушением поверхностей нагрева паровых котлов. Диаметр молекулы водорода 0,212, диз метр атома водорода 0,1 нм. Энергия диссоциации молекулы водорода на атомы составляет 432 Дж/моль (103 ккал/моль). Термическая диссоциация молекул Нг при температурах ниже 2000°С очень глала при 225 °С константа диссоциации /Сн.=Ю . [c.20]
АН298 = 2500 2000 кал/моль (среднее из величин 3200 2000 и 1800 2000 кал/моль, рассчитанных соответственно по II и III законам). Используя далее для теллура AH ag 39 600 [39] и для Тег (г) Do = 62 300 2000 кал/моль [106], они получили для энергии диссоциации молекулы ТеО Do = 92 500 2000 кал/моль, что подтверждает спектроскопические оценки. Величина Муэнова должна считаться в настоящее время наиболее надежной. Для энергии атомизации димера (ТеО)г в работе [119] получено [c.81]
СеЗе. Состав пара моноселенида германия хорошо изучен [159, 160], испарение его происходит в виде простейших двухатомных молекул. Химический и рентгенофазовый анализы показывают, что ОеЗе возгоняется без изменения состава [161]. Довольно высокая энергия диссоциации молекулы ОеЗе практически исключает возможность диссоциации соединения в паре при температурах экспериментов. [c.86]
Теплота и изобарный потенциал образования ТеО и-(ТеО) 2- В справочнике [40] приводятся величины АЯ 298 = 41 563 и AG/298 = 35 252 кал/моль, основанные на заниженном значении энергии диссоциации молекулы ТеО. Муэнов и др. [119] на основании своего значения Do рассчитали для ТеО (г) ДЯ/298 = 17 300 2000 кал/моль. Эта величина рекомендуется в настоящем справочнике. Пользуясь принятыми в этом справочнике значениями S298, для изобарного потенциала образования ТеО (г) получаем AG°fm =11 ООО 2000 кал/моль. [c.87]
Энергия диссоциации. Согласно спектроскопическим [231] и масс-спектрометрическим [232] данным, энергия диссоциации молекулы 5ЬТе О = 2,6 эв, или 60 378 2400 кал/моль [50]. [c.100]
На основе молекулярно-орбитальной теории также трудно простым образом объяснить, почему молекулы типа НгР, или Н О или Л Н4 не существуют или имеют сравнительно малые энергии диссоциации на Н + НР, Н Н Н2О и Н + КНз. Другими словами, трудно объяснить, почему валентность атома Р насыщается одним атомом Н, валентность О — двумя атомами Н и валентность атома N — тремя атомами Н. Если на орбитали, изображенные на диаграмме фиг. 123, помещается девять электронов, то иослед-ний электрон должен попасть на разрыхляющую орбиталь 4 , так что в итоге останутся только три связывающих электрона. Из этих соображений следует, что молекула НаЕ, возможно, будет стабильной однако стабильность такой молекулы будет существенно меньшей, чем стабильность молекулы Н2О, прежде всего за счет того, что орбиталь 4а( является сильно разрыхляющей орбиталью, коррелирующей с орбиталью объединенного атома, главное квантовое число которой равно 3. На основе этих соображений следует ожидать малой величины энергии диссоциации молекулы НзЕ на Н и НР. До сих пор не было получено никаких данных о существовании радикала НгР- Тем не менее были получены полимеры НР, [c.411]
При малой диссоциации а < 1 изменение давления невелико, но изменение энерпр и теплоемкости газа все равно может оказаться значительным. Пусть 8а2 — энергия одной молекулы при температуре Г, а ед — энергия одного атома. Обозначим энергию, необходимую для разрыва невозбужденной молекулы (т. е. в отсутствие врапцений и колебаний или при Т = Ь) через и. V представляет собой энергию связи или энергию диссоциации молекулы например, для Ог и = 5,11 эв 118 ккал/моль ), и/к = 59 400° К N3 V э — «22Ъ ккал/моль, и/к = ИЗ 000° К [c.159]
Теплоты реакций здесь соответствуют энергиям диссоциации молекул N2 и N0, равным 9,74 эв = 225 ккал1молъ и 6,5 эв = 150 ккал/моль ). [c.320]
Совсем иначе обстоит дело при образовании слабосвязанного состояния с 8 = 2 эВ, когда освобождающейся энергии не хватает на ионизацию электрона. Другой процесс передачи энергии—диссоциация молекулы 1>2 — также требует большой энергии (около 4,5 эВ). Поэтому единственно возможным процессом перёдачи энергии является возбуждение колебательных уровней в системе расстояние между которыми мало (А.Еосц 0,3 эВ). Точный расчет показал, что [c.191]
c.66
]
Теплотехнический справочник Том 1 (1957) — [
c.66
]
From Wikipedia, the free encyclopedia
The bond-dissociation energy (BDE, D0, or DH°) is one measure of the strength of a chemical bond A−B. It can be defined as the standard enthalpy change when A−B is cleaved by homolysis to give fragments A and B, which are usually radical species.[1][2] The enthalpy change is temperature-dependent, and the bond-dissociation energy is often defined to be the enthalpy change of the homolysis at 0 K (absolute zero), although the enthalpy change at 298 K (standard conditions) is also a frequently encountered parameter.[3]
As a typical example, the bond-dissociation energy for one of the C−H bonds in ethane (C2H6) is defined as the standard enthalpy change of the process
- CH3CH2−H → CH3CH2• + H•,
- DH°298(CH3CH2−H) = ΔH° = 101.1(4) kcal/mol = 423.0 ± 1.7 kJ/mol = 4.40(2) eV (per bond).[4]
To convert a molar BDE to the energy needed to dissociate the bond per molecule, the conversion factor 23.060 kcal/mol (96.485 kJ/mol) for each eV can be used.
A variety of experimental techniques, including spectrometric determination of energy levels, generation of radicals by pyrolysis or photolysis, measurements of chemical kinetics and equilibrium, and various calorimetric and electrochemical methods have been used to measure bond dissociation energy values. Nevertheless, bond dissociation energy measurements are challenging and are subject to considerable error. The majority of currently known values are accurate to within ±1 or 2 kcal/mol (4–10 kJ/mol).[5] Moreover, values measured in the past, especially before the 1970s, can be especially unreliable and have been subject to revisions on the order of 10 kcal/mol (e.g., benzene C–H bonds, from 103 kcal/mol in 1965 to the modern accepted value of 112.9(5) kcal/mol). Even in modern times (between 1990 and 2004), the O−H bond of phenol has been reported to be anywhere from 85.8 to 91.0 kcal/mol.[6] On the other hand, the bond dissociation energy of H2 at 298 K has been measured to high precision and accuracy: DH°298(H−H) = 104.1539(1) kcal/mol or 435.780 kJ/mol.[5]
Definitions and related parameters[edit]
The term bond-dissociation energy is similar to the related notion of bond-dissociation enthalpy (or bond enthalpy), which is sometimes used interchangeably. However, some authors make the distinction that the bond-dissociation energy (D0) refers to the enthalpy change at 0 K, while the term bond-dissociation enthalpy is used for the enthalpy change at 298 K (unambiguously denoted DH°298). The former parameter tends to be favored in theoretical and computational work, while the latter is more convenient for thermochemical studies. For typical chemical systems, the numerical difference between the quantities is small, and the distinction can often be ignored. For a hydrocarbon RH, where R is significantly larger than H, for instance, the relationship D0(R−H) ≈ DH°298(R−H) − 1.5 kcal/mol is a good approximation.[7] Some textbooks ignore the temperature dependence,[8] while others have defined the bond-dissociation energy to be the reaction enthalpy of homolysis at 298 K.[9][10][11]
The bond dissociation energy is related to but slightly different from the depth of the associated potential energy well of the bond, De, known as the electronic energy. This is due to the existence of a zero-point energy ε0 for the vibrational ground state, which reduces the amount of energy needed to reach the dissociation limit. Thus, D0 is slightly less than De, and the relationship D0 = De − ε0 holds.[7]
The bond dissociation energy is an enthalpy change of a particular chemical process, namely homolytic bond cleavage, and «bond strength» as measured by the BDE should not be regarded as an intrinsic property of a particular bond type but rather as an energy change that depends on the chemical context. For instance, Blanksby and Ellison cites the example of ketene (H2C=CO), which has a C=C bond dissociation energy of 79 kcal/mol, while ethylene (H2C=CH2) has a bond dissociation energy of 174 kcal/mol. This vast difference is accounted for by the thermodynamic stability of carbon monoxide (CO), formed upon the C=C bond cleavage of ketene.[7] The difference in availability of spin states upon fragmentation further complicates the use of BDE as a measure of bond strength for head-to-head comparisons, and force constants have been suggested as an alternative.[12]
Historically, the vast majority of tabulated bond energy values are bond enthalpies. More recently, however, the free energy analogue of bond-dissociation enthalpy, known as the bond-dissociation free energy (BDFE), has become more prevalent in the chemical literature. The BDFE of a bond A–B can be defined in the same way as the BDE as the standard free energy change (ΔG°) accompanying homolytic dissociation of AB into A and B. However, it is often thought of and computed stepwise as the sum of the free-energy changes of heterolytic bond dissociation (A–B → A+ + :B−), followed by one-electron reduction of A (A+ + e− → A•) and one-electron oxidation of B (:B− → •B + e−).[13] In contrast to the BDE, which is usually defined and measured in the gas phase, the BDFE is often determined in the solution phase with respect to a solvent like DMSO, since the free-energy changes for the aforementioned thermochemical steps can be determined from parameters like acid dissociation constants (pKa) and standard redox potentials (ε°) that are measured in solution.[14]
Bond energy[edit]
Except for diatomic molecules, the bond-dissociation energy differs from the bond energy. While the bond-dissociation energy is the energy of a single chemical bond, the bond energy is the average of all the bond-dissociation energies of the bonds of the same type for a given molecule.[15] For a homoleptic compound EXn, the E–X bond energy is (1/n) multiplied by the enthalpy change of the reaction EXn → E + nX. Average bond energies given in tables are the average values of the bond energies of a collection of species containing «typical» examples of the bond in question.
For example, dissociation of HO−H bond of a water molecule (H2O) requires 118.8 kcal/mol (497.1 kJ/mol). The dissociation of the remaining hydroxyl radical requires 101.8 kcal/mol (425.9 kJ/mol). The bond energy of the covalent O−H bonds in water is said to be 110.3 kcal/mol (461.5 kJ/mol), the average of these values.[16]
In the same way, for removing successive hydrogen atoms from methane the bond-dissociation energies are 105 kcal/mol (439 kJ/mol) for D(CH3−H), 110 kcal/mol (460 kJ/mol) for D(CH2−H), 101 kcal/mol (423 kJ/mol) for D(CH−H) and finally 81 kcal/mol (339 kJ/mol) for D(C−H). The bond energy is, thus, 99 kcal/mol, or 414 kJ/mol (the average of the bond-dissociation energies). None of the individual bond-dissociation energies equals the bond energy of 99 kcal/mol.[17][7]
Strongest bonds and weakest bonds[edit]
According to BDE data, the strongest single bonds are Si−F bonds. The BDE for H3Si−F is 152 kcal/mol, almost 50% stronger than the H3C−F bond (110 kcal/mol). The BDE for F3Si−F is even larger, at 166 kcal/mol. One consequence to these data are that many reactions generate silicon fluorides, such as glass etching, deprotection in organic synthesis, and volcanic emissions.[18] The strength of the bond is attributed to the substantial electronegativity difference between silicon and fluorine, which leads to a substantial contribution from both ionic and covalent bonding to the overall strength of the bond.[19] The C−C single bond of diacetylene (HC≡C−C≡CH) linking two sp-hybridized carbon atoms is also among the strongest, at 160 kcal/mol.[5] The strongest bond for a neutral compound, including multiple bonds, is found in carbon monoxide at 257 kcal/mol. The protonated forms of CO, HCN and N2 are said to have even stronger bonds, although another study argues that the use of BDE as a measure of bond strength in these cases is misleading.[12]
On the other end of the scale, there is no clear boundary between a very weak covalent bond and an intermolecular interaction. Lewis acid–base complexes between transition metal fragments and noble gases are among the weakest of bonds with substantial covalent character, with (CO)5W:Ar having a W–Ar bond dissociation energy of less than 3.0 kcal/mol.[20] Held together entirely by the van der Waals force, helium dimer, He2, has the lowest measured bond dissociation energy of only 0.021 kcal/mol.[21]
Homolytic versus heterolytic dissociation[edit]
Bonds can be broken symmetrically or asymmetrically. The former is called homolysis and is the basis of the usual BDEs. Asymmetric scission of a bond is called heterolysis. For molecular hydrogen, the alternatives are:
-
Symmetric: H2 → 2 H• ΔH° = 104.2 kcal/mol (see table below) Asymmetric: H2 → H+ + H− ΔH° = 400.4 kcal/mol (gas phase)[22] Asymmetric: H2 → H+ + H− ΔG° = 34.2 kcal/mol (in water)[23] (pKaaq = 25.1)
In the gas phase, the enthalpy of heterolysis is larger than that of homolysis, due to the need to separate unlike charges. However, this value is lowered substantially in the presence of a solvent.
Representative bond enthalpies[edit]
The data tabulated below shows how bond strengths vary over the periodic table.
| Bond | Bond | Bond-dissociation enthalpy at 298 K | Comment | ||
|---|---|---|---|---|---|
| (kcal/mol) | (kJ/mol) | (eV/bond) | |||
| C−C | in typical alkane | 83–90 | 347–377 | 3.60–3.90 | Strong, but weaker than C−H bonds |
| C−F | in CH3F | 115 | 481 | 4.99 | Very strong, rationalizes inertness of Teflon |
| C−Cl | in CH3Cl | 83.7 | 350 | 3.63 | Strong, but considerably weaker than C−F bonds |
| F−F | fluorine | 37 | 157 | 1.63 | Very weak, in conjunction with strong C−F and H−F bonds, leads to an explosive reaction with hydrocarbons |
| Cl−Cl | chlorine | 58 | 242 | 2.51 | Indicated by facility of photochemical chlorinations |
| Br−Br | bromine | 46 | 192 | 1.99 | Indicated by facility of photochemical brominations |
| I−I | iodine | 36 | 151 | 1.57 | Indicated by catalysis of cis/trans isomerization |
| H−H | hydrogen | 103 | 431 | 4.52 | Strong, nonpolarizable bond |
| H−F | hydrogen fluoride | 136 | 569 | 5.90 | Very strong |
| O−H | in water | 119 | 497 | 5.15 | Very strong, hydroxyl radical reactive with almost all organics exothermically by H atom abstraction |
| O−H | in methanol | 105 | 440 | 4.56 | Slightly stronger than C−H bonds |
| O−H | in α-tocopherol (an antioxidant) | 77 | 323 | 3.35 | O−H bond strength depends strongly on substituent on O |
| C-O | methanol | 92 | 385 | 3.99 | typical alcohol |
| C≡O | carbon monoxide | 257 | 1077 | 11.16 | Strongest bond in neutral molecule |
| O=CO | carbon dioxide | 127 | 532 | 5.51 | Slightly stronger than C−H bonds, surprisingly low due to stability of C≡O |
| O=CH2 | formaldehyde | 179 | 748 | 7.75 | Much stronger than C−H bonds |
| O=O | oxygen | 119 | 498 | 5.15 | Stronger than single bonds, weaker than many other double bonds |
| N≡N | nitrogen | 226 | 945 | 9.79 | One of the strongest bonds, large activation energy in production of ammonia |
There is great interest, especially in organic chemistry, concerning relative strengths of bonds within a given group of compounds, and representative bond dissociation energies for common organic compounds are shown below.[7][17]
| Bond | Bond | Bond-dissociation energy at 298 K | Comment | ||
|---|---|---|---|---|---|
| (kcal/mol) | (kJ/mol) | (eV/bond) | |||
| H3C−H | Methyl C−H bond | 105 | 439 | 4.550 | One of the strongest aliphatic C−H bonds |
| C2H5−H | Ethyl C−H bond | 101 | 423 | 4.384 | Slightly weaker than H3C−H |
| (CH3)2CH−H | Isopropyl C−H bond | 99 | 414 | 4.293 | Secondary radicals are stabilized |
| (CH3)3C−H | t-Butyl C−H bond | 96.5 | 404 | 4.187 | Tertiary radicals are even more stabilized |
| (CH3)2NCH2−H | C−H bond α to amine | 91 | 381 | 3.949 | Lone-pair bearing heteroatoms weaken C−H bonds |
| (CH2)3OCH−H | C−H bond α to ether | 92 | 385 | 3.990 | Lone-pair bearing heteroatoms weaken C−H bonds. THF tends to form hydroperoxides |
| CH3C(=O)CH2−H | C−H bond α to ketone | 96 | 402 | 4.163 | Conjugating electron-withdrawing groups weaken C−H bonds |
| CH2CH−H | Vinyl C−H bond | 111 | 464 | 4.809 | Vinyl radicals are uncommon |
| HCC−H | Acetylenic C−H bond | 133 | 556 | 5.763 | Acetylenic radicals are very rare |
| C6H5−H | Phenyl C−H bond | 113 | 473 | 4.902 | Comparable to vinyl radical, uncommon |
| CH2CHCH2−H | Allylic C−H bond | 89 | 372 | 3.856 | Such bonds show enhanced reactivity, see drying oil |
| C6H5CH2−H | Benzylic C−H bond | 90 | 377 | 3.907 | Akin to allylic C−H bonds. Such bonds show enhanced reactivity |
| H3C−CH3 | Alkane C−C bond | 83–90 | 347–377 | 3.60–3.90 | Much weaker than C−H bond. Homolytic cleavage occurs when H3C−CH3 thermolysed at >500 °C |
| H2C=CH2 | Alkene C=C bond | ~170 | ~710 | ~7.4 | About 2 times stronger than a C−C single bond; however, the π bond (~65 kcal/mol) is weaker than the σ bond |
| HC≡CH | Alkyne C≡C triple bond | ~230 | ~960 | ~10.0 | About 2.5 times stronger than a C−C single bond |
See also[edit]
- Bond energy
- Electronegativity
- Ionization energy
- Electron affinity
- Lattice energy
References[edit]
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the «Gold Book») (1997). Online corrected version: (2006–) «Bond-dissociation energy». doi:10.1351/goldbook.B00699
- ^ The value reported as the bond-dissociation energy (BDE) is generally the enthalpy of the homolytic dissociation of a gas-phase species. For instance, the BDE of diiodine is calculated as twice the heat of formation of iodine radical (25.5 kcal/mol) minus the heat of formation of diiodine gas (14.9 kcal/mol). This gives the accepted BDE of diiodine of 36.1 kcal/mol. (By definition, diiodine in the solid state has a heat of formation of 0.)
- ^ The IUPAC Gold Book does not stipulate a temperature for its definition of bond-dissociation energy (ref. 1).
- ^ The corresponding BDE at 0 K (D0) is 99.5(5) kcal/mol.
- ^ a b c Luo, Y. R. (2007). Comprehensive handbook of chemical bond energies. Boca Raton: CRC Press. ISBN 978-0-8493-7366-4. OCLC 76961295.
- ^ Mulder P., Korth H. G., Pratt D. A., DiLabio G. A., Valgimigli L., Pedulli G. F., Ingold K. U. (March 2005). «Critical re-evaluation of the O−H bond dissociation enthalpy in phenol». The Journal of Physical Chemistry A. 109 (11): 2647–55. Bibcode:2005JPCA..109.2647M. doi:10.1021/jp047148f. PMID 16833571.
{{cite journal}}: CS1 maint: uses authors parameter (link) - ^ a b c d e Blanksby S. J., Ellison G. B. (April 2003). «Bond dissociation energies of organic molecules». Accounts of Chemical Research. 36 (4): 255–63. CiteSeerX 10.1.1.616.3043. doi:10.1021/ar020230d. PMID 12693923.
{{cite journal}}: CS1 maint: uses authors parameter (link) - ^ Anslyn, Eric V.; Dougherty, Dennis A. (2006). Modern physical organic chemistry. Sausalito, CA: University Science. ISBN 978-1-891389-31-3. OCLC 55600610.
- ^ Darwent, B. deB. (January 1970). Bond Dissociation Energies in Simple Molecules (PDF). NSRDS-NBS 31. Washington, DC: U.S. National Bureau of Standards. LCCN 70602101.
- ^ Streitwieser, Andrew; Heathcock, Clayton H.; Kosower, Edward M. (2017). Introduction to Organic Chemistry. New Delhi: Medtech (Scientific International, reprint of 4th revised edition, 1998, Macmillan). p. 101. ISBN 9789385998898.
- ^ Carroll, Felix A. (2010). Perspectives on structure and mechanism in organic chemistry (2nd ed.). Hoboken, N.J.: John Wiley. ISBN 978-0-470-27610-5. OCLC 286483846.
- ^ a b Kalescky, Robert; Kraka, Elfi; Cremer, Dieter (2013-08-30). «Identification of the Strongest Bonds in Chemistry». The Journal of Physical Chemistry A. 117 (36): 8981–8995. Bibcode:2013JPCA..117.8981K. doi:10.1021/jp406200w. ISSN 1089-5639. PMID 23927609. S2CID 11884042.
- ^ Miller D. C., Tarantino K. T., Knowles R. R. (June 2016). «Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities». Topics in Current Chemistry. 374 (3): 30. doi:10.1007/s41061-016-0030-6. PMC 5107260. PMID 27573270.
{{cite journal}}: CS1 maint: uses authors parameter (link) - ^ Bordwell, F. G.; Cheng, Jin Pei; Harrelson, John A. (February 1988). «Homolytic bond dissociation energies in solution from equilibrium acidity and electrochemical data». Journal of the American Chemical Society. 110 (4): 1229–1231. doi:10.1021/ja00212a035.
- ^ Norman, Richard O. C.; Coxon, James M. (2001). Principles of organic synthesis (3rd ed.). London: Nelson Thornes. p. 7. ISBN 978-0-7487-6162-3. OCLC 48595804.
- ^ Lehninger, Albert L.; Nelson, David L.; Cox, Michael M. (2005). Lehninger Principles of Biochemistry (4th ed.). W. H. Freeman. p. 48. ISBN 978-0-7167-4339-2. Retrieved May 20, 2016.
- ^ a b Streitwieser A.; Bergman R. G. (19 September 2018). «Table of Bond Dissociation Energies». University of California, Berkeley. Retrieved 13 March 2019.
- ^ Lide, David R., ed. (2006). CRC Handbook of Chemistry and Physics (87th ed.). Boca Raton, FL: CRC Press. ISBN 0-8493-0487-3.
- ^ Gillespie, Ronald J. (July 1998). «Covalent and Ionic Molecules: Why Are BeF2 and AlF3 High Melting Point Solids whereas BF3 and SiF4 Are Gases?». Journal of Chemical Education. 75 (7): 923. Bibcode:1998JChEd..75..923G. doi:10.1021/ed075p923. ISSN 0021-9584.
- ^ Grills D. C.; George M. W. (2001), «Transition metal-noble gas complexes», Advances in Inorganic Chemistry, Elsevier, pp. 113–150, doi:10.1016/s0898-8838(05)52002-6, ISBN 9780120236527.
- ^ Cerpa, Erick; Krapp, Andreas; Flores-Moreno, Roberto; Donald, Kelling J.; Merino, Gabriel (2009-02-09). «Influence of Endohedral Confinement on the Electronic Interaction between He atoms: A He2@C20H20 Case Study». Chemistry – A European Journal. 15 (8): 1985–1990. doi:10.1002/chem.200801399. ISSN 0947-6539. PMID 19021178.
- ^ Bartmess, John E.; Scott, Judith A.; McIver, Robert T. (September 1979). «Scale of acidities in the gas phase from methanol to phenol». Journal of the American Chemical Society. 101 (20): 6046–6056. doi:10.1021/ja00514a030.
- ^ Connelly, Samantha J.; Wiedner, Eric S.; Appel, Aaron M. (2015-03-17). «Predicting the reactivity of hydride donors in water: thermodynamic constants for hydrogen». Dalton Transactions. 44 (13): 5933–5938. doi:10.1039/C4DT03841J. ISSN 1477-9234. PMID 25697077.
Энергия диссоциации связи (BDE, D0или DH ° ) — это один из показателей прочности химической связи A – B. Его можно определить как стандартное изменение энтальпии, когда A – B расщепляется в результате гомолиза с образованием фрагментов A и B, которые обычно являются радикалами. Изменение энтальпии зависит от температуры, и энергия диссоциации связи часто определяется как изменение энтальпии гомолиза при 0 K (абсолютном нуле ), хотя изменение энтальпии при 298 K (стандартные условия ) также является часто встречающимся параметром. В качестве типичного примера энергия диссоциации одной из связей C − H в этане (C2H6) определяется как стандартное изменение энтальпии процесса
- CH3CH2–H → CH3CH2 + H,
- DH ° 298 (CH 3CH2-H) = ΔH ° = 101,1 (4) ккал / моль = 423,0 ± 1,7 кДж / моль = 4,40 (2) эВ (на связь).
Чтобы преобразовать молярный БДЭ в энергию, необходимую для диссоциации связи на молекулу, коэффициент преобразования 23,060 ккал / моль (96,485 кДж / моль) для каждого эВ может быть используемый.
Разнообразные экспериментальные методы, включая спектрометрическое определение уровней энергии, генерацию радикалов с помощью пиролиза или фотолиза, измерения химической кинетики и равновесие и различные калориметрические и электрохимические методы были использованы для измерения значений энергии диссоциации связи. Тем не менее, измерение энергии диссоциации связи является сложной задачей и может содержать значительные ошибки. Большинство известных в настоящее время значений имеют точность в пределах ± 1 или 2 ккал / моль (4–10 кДж / моль). Более того, значения, измеренные в прошлом, особенно до 1970-х годов, могут быть особенно ненадежными и подвергались пересмотру порядка 10 ккал / моль (например, бензольные связи C – H, с 103 ккал / моль в 1965 году до современных принятое значение 112,9 (5) ккал / моль). Даже в наше время (между 1990 и 2004 годами) связь O-H фенола составляла от 85,8 до 91,0 ккал / моль. С другой стороны, энергия диссоциации связи H 2 при 298 K была измерена с высокой точностью и точностью: DH ° 298 (H-H) = 104,1539 (1) ккал.
Определения и связанные параметры
Термин энергия диссоциации связи аналогичен соответствующему понятию энтальпии диссоциации связи (или энтальпии связи), которое иногда используется взаимозаменяемо. Однако некоторые авторы делают различие, что энергия диссоциации связи (D 0) относится к изменению энтальпии при 0 K, в то время как термин энтальпия диссоциации связи используется для изменения энтальпии при 298 K (однозначно обозначено DH ° 298). Первый параметр, как правило, предпочитается в теоретической и вычислительной работе, в то время как второй более удобен для термохимических исследований. Для типичных химических систем численное различие между величинами невелико, и различие часто можно игнорировать. Например, для углеводородного RH, где R значительно больше H, соотношение D 0 (R-H) ≈ DH ° 298 (R-H) — 1,5 ккал / моль — хорошее приближение. Некоторые учебники игнорируют температурную зависимость, в то время как другие определяют энергию диссоциации связи как энтальпию реакции гомолиза при 298 К.
Энергия диссоциации связи связана с глубиной связанного потенциальная энергетическая яма связи, D e, известная как электронная энергия. Это связано с существованием энергии нулевой точки ε0для основного колебательного состояния, что снижает количество энергии, необходимое для достижения предела диссоциации. Таким образом, D 0 немного меньше, чем D e, и соотношение D 0 = D e — ε 0.
Энергия диссоциации связи — это изменение энтальпии конкретного химического процесса, а именно гомолитического разрыва связи, и «прочность связи», измеренная с помощью BDE, не должна рассматриваться как внутреннее свойство конкретного тип связи, а скорее как изменение энергии, которое зависит от химического контекста. Например, Бланксби и Эллисон приводят пример кетена (H 9 2 76 C = CO), у которого энергия диссоциации связи C = C составляет 79 ккал / моль, а этилен (H 9 2 C = CH 2) имеет энергию диссоциации связи 174 ккал / моль. Это огромное различие объясняется термодинамической стабильностью монооксида углерода (CO), образующегося при разрыве связи C = C кетена. Различие в доступности спиновых состояний при фрагментации еще больше усложняет использование БДЭ в качестве меры прочности связи для сравнений «голова к голове», и в качестве альтернативы были предложены силовые константы.
Исторически сложилось так, что подавляющее большинство табличных значений энергии связи представляют собой энтальпии связи. Однако в последнее время в химической литературе все более распространенным стал аналог свободной энергии энтальпии диссоциации связи, известный как свободная энергия диссоциации связи (BDFE). BDFE связи A – B можно определить так же, как BDE, как стандартное изменение свободной энергии (ΔG °), сопровождающее гомолитическую диссоциацию AB на A и B. Однако его часто считают и вычисляют пошагово как сумма изменений свободной энергии при диссоциации гетеролитической связи (A – B → A +: B) с последующим одноэлектронным восстановлением A (A + e → A •) и одноэлектронным окислением B (: B → • В + д). В отличие от BDE, который обычно определяется и измеряется в газовой фазе, BDFE часто определяется в фазе раствора по отношению к растворителю, подобному DMSO, поскольку изменения свободной энергии для вышеупомянутых термохимических стадий могут быть определены из параметров например, константы диссоциации кислоты (pK a) и стандартные окислительно-восстановительные потенциалы (°), которые измеряются в растворе.
Энергия связи
За исключением двухатомных молекул, энергия диссоциации связи отличается от энергии связи . В то время как энергия диссоциации связи — это энергия одинарной химической связи, энергия связи — это среднее значение всех энергий диссоциации связей одного типа для данной молекулы. Для гомолептического соединения EX n энергия связи E – X равна (1 / n), умноженному на изменение энтальпии реакции EX n → E + nX. Средние энергии связи, приведенные в таблицах, представляют собой средние значения энергий связи для набора разновидностей, содержащих «типичные» примеры рассматриваемой связи.
Например, для диссоциации связи HO −H молекулы воды (H 2 O) требуется 118,8 ккал / моль (497,1 кДж / моль). Для диссоциации оставшегося гидроксильного радикала требуется 101,8 ккал / моль (425,9 кДж / моль). Энергия связи ковалентных связей O −H в воде составляет 110,3 ккал / моль (461,5 кДж / моль), среднее из этих значений.
Таким же образом для удаления следующих друг за другом атомов водорода из метана, энергии диссоциации связи составляют 105 ккал / моль (439 кДж / моль) для D (CH 3 -H), 110 ккал / моль (460 кДж / моль) для D (CH 2 -H), 101 ккал / моль (423 кДж / моль) для D (CH-H) и, наконец, 81 ккал / моль (339 кДж / моль) для D (C-H). Таким образом, энергия связи составляет 99 ккал / моль или 414 кДж / моль (среднее значение энергии диссоциации связи). Ни одна из индивидуальных энергий диссоциации связи не равна энергии связи 99 ккал / моль.
Самые сильные и самые слабые связи
Согласно данным BDE, самые сильные одинарные связи — это связи Si-F. БДЭ для H 3 Si-F составляет 152 ккал / моль, что почти на 50% сильнее, чем связь H 3 C-F (110 ккал / моль). БДЭ для F 3 Si-F еще больше — 166 ккал / моль. Одним из следствий этих данных является то, что во многих реакциях образуются фториды кремния, такие как травление стекла, снятие защиты в органическом синтезе и вулканические выбросы. Прочность связи объясняется значительной разницей в электроотрицательности кремния и фтора, что приводит к значительному вкладу ионной и ковалентной связи в общую прочность связи. Простая связь C-C в диацетилене (HC≡C-C≡CH), связывающая два sp-гибридизированных атома углерода, также является одной из самых прочных, при 160 ккал / моль. Самая прочная связь для нейтрального соединения, включая кратные связи, обнаруживается в монооксиде углерода при 257 ккал / моль. Считается, что протонированные формы CO, HCN и N 2 имеют даже более прочные связи, хотя другое исследование утверждает, что использование БДЭ в качестве меры прочности связи в этих случаях вводит в заблуждение.
На другом конце шкалы нет четкой границы между очень слабой ковалентной связью и межмолекулярным взаимодействием. Кислотно-основные комплексы Льюиса между фрагментами переходных металлов и благородными газами являются одними из самых слабых из связей с существенно ковалентным характером, при этом (CO) 5 W: Ar, имеющий энергию диссоциации связи W-Ar менее 3,0 ккал. / моль. Удерживаемый полностью силой Ван-дер-Ваальса, димер гелия, He 2, имеет самую низкую измеренную энергию диссоциации связи, составляющую всего 0,021 ккал / моль.
Гомолитическая диссоциация в сравнении с гетеролитической
Связи могут быть разорваны симметрично или асимметрично. Первый называется гомолизом и является основой обычных БДЭ. Асимметричный разрыв связи называется гетеролизом. Для молекулярного водорода альтернативы:
-
Симметричный: H2→ 2 H ΔH ° = 104,2 ккал / моль (см. Таблицу ниже) Асимметричный: H2→ H + H ΔH ° = 400,4 ккал / моль (газовая фаза) Асимметричный: H2→ H + H ΔG ° = 34,2 ккал / моль (в воде) (pK a = 25,1)
Обратите внимание, что в газовой фазе энтальпия гетеролиза больше, чем энтальпия гомолиза, из-за необходимости разделять разноименные заряды. Однако это значение существенно снижается в присутствии растворителя.
Данные, представленные в таблице ниже, показывают, как сила сцепления меняется по периодической таблице.
| Связь | Связь | Энергия диссоциации связи при 298 K | Комментарий | ||
|---|---|---|---|---|---|
| (ккал / моль) | (кДж / моль) | (эВ / связь) | |||
| C − C | в типичном алкане | 83–90 | 347–377 | 3,60–3,90 | Сильные, но более слабые, чем связи C − H |
| C − F | в CH 3F | 115 | 481 | 4,99 | Очень прочный, рационализирует инертность тефлона |
| C-Cl | в CH 3 Cl | 83,7 | 350 | 3,63 | Сильные, но значительно более слабые, чем связи C-F |
| F-F | фтор | 37 | 157 | 1,63 | Очень слабый, в сочетании с сильными связями C-F и H-F, приводит к взрывной реакции с углеводородами |
| Cl-Cl | хлор | 58 | 242 | 2,51 | Обозначено установкой фотохимического хлорирования |
| Br-Br | бром | 46 | 192 | 1,99 | Обозначается установкой фотохимического бромирования |
| I-I | йод | 36 | 151 | 1,57 | Обнаруживается катализом цис / транс-изомеризация |
| H-H | водород | 104 | 436 | 4,52 | Сильная неполяризуемая связь |
| H-F | фтороводород | 136 | 569 | 5.90 | Очень сильное |
| O-H | в воде | 119 | 497 | 5.15 | Очень сильный, гидроксильный радикал реакционноспособен почти со всеми органическими соединениями экзотермически за счет отрыва атома H |
| O-H | в метаноле | 105 | 440 | 4,56 | Немного прочнее, чем связи C-H |
| O-H | в α- токофероле ( антиоксидант) | 77 | 323 | 3,35 | Прочность связи O-H сильно зависит от заместителя на атоме углерода O |
| C≡O | монооксид | 257 | 1077 | 11,16 | Самая сильная связь в нейтральной молекуле |
| O = CO | диоксид углерода | 127 | 532 | 5,51 | Немного прочнее, чем связи C-H, удивительно низкое из-за стабильности C≡O |
| O = CH 2 | формальдегид | 179 | 748 | 7,75 | Намного прочнее, чем связи C-H |
| O = O | кислород | 119 | 498 | 5.15 | Сильнее n одинарных связей, более слабых, чем многие другие двойные связи |
| N≡N | азот | 226 | 945 | 9,79 | Одна из самых прочных связей, большая энергия активации при производстве аммиака |
Существует большой интерес, особенно в органической химии, относительно относительной силы связей внутри данной группы соединений и типичных энергий диссоциации связей для обычных органических соединений показаны ниже.
| Связь | Связь | Энергия диссоциации связи при 298 K | Комментарий | ||
|---|---|---|---|---|---|
| (ккал / моль) | ( кДж / моль) | (эВ / связь) | |||
| H3C-H | Метил C-H-связь | 105 | 439 | 4,550 | Одна из самых сильных алифатических связей C − H |
| C2H5-H | Этил Связь C − H | 101 | 423 | 4,384 | Немного слабее, чем H 3 C-H |
| (CH 3)2CH-H | Изопропил C-H связь | 99 | 414 | 4,293 | Вторичные радикалы стабилизированы |
| (CH 3)3C-H | трет-Бутил C- H связь | 96,5 | 404 | 4,187 | Третичный рад соединения еще более стабилизированы |
| (CH 3)2NCH 2 -H | C-H связь α с амином | 91 | 381 | 3.949 | Гетероатомы, несущие неподеленную пару, ослабляют связи C − H |
| (CH 2)3OCH − H | связь C − H α с эфиром | 92 | 385 | 3.990 | Гетероатомы, несущие неподеленную пару, ослабляют связи C-H. ТГФ имеет тенденцию к образованию гидропероксидов |
| CH3C (= O) CH 2 -H | C-H-связь α с кетоном | 96 | 402 | 4,163 | Конъюгированные электроноакцепторные группы ослабляют связи C − H |
| CH2CH − H | Винил связь C − H | 111 | 464 | 4.809 | Виниловые радикалы встречаются редко |
| HCC-H | Ацетиленовая связь C-H | 133 | 556 | 5,763 | Ацетиленовые радикалы очень редки |
| C6H5-H | Фенил связь C-H | 113 | 473 | 4,902 | Сравнимо с виниловым радикалом, редко |
| CH2CHCH 2 -H | Аллильная связь C-H | 89 | 372 | 3,856 | Такие связи проявляют повышенную реакционную способность, см. олифа |
| C6H5CH2-H | Бензильная связь C-H | 90 | 377 | 3.907 | Родственны аллильным связям C-H. Такие связи обладают повышенной реакционной способностью |
| H3C-CH 3 | Алкан Связь C-C | 83–90 | 347–377 | 3,60–3,90 | Намного слабее, чем связь C − H. Гомолитическое расщепление происходит при термолизе H 3 C-CH 3 при>500 ° C |
| H2C = CH 2 | Алкен C = C-связь | ~ 170 | ~ 710 | ~ 7.4 | Примерно в 2 раза прочнее, чем одинарная связь C − C; однако π-связь (~ 65 ккал / моль) слабее, чем σ-связь |
| HC≡CH | Алкин C≡C тройная связь | ~ 230 | ~ 960 | ~ 10,0 | Примерно в 2,5 раза сильнее, чем одинарная связь C − C |
См. Также
Ссылки
Энергия — диссоциация — двухатомная молекула
Cтраница 1
Энергия диссоциации двухатомной молекулы равна энергии связи между атомами в этой молекуле.
[1]
Энергии диссоциации двухатомных молекул определяются на основании данных анализа молекулярных спектров.
[2]
Энергии диссоциации двухатомных молекул часто могут быть измерены и использованы для вычисления теплот других реакций.
[3]
Энергии диссоциации двухатомных молекул закономерно зависят от природы химической связи. При одинаковых анионах замена металлов а-подгрупп их — аналогами также приводит к уменьшению D. Вместе с тем, изменение валентности металлов мало влияет на энергию диссоциации двухатомных молекул. Количественная интерпретация этого факта будет дана в дальнейшем изложении, а пока остановимся на качественной стороне явления.
[4]
Энергии диссоциации двухатомных молекул определяются на основании данных анализа молекулярных спектров.
[5]
Энергия диссоциации двухатомных молекул равна 1 78 эв и теплота испарения — 95 4 ккал / молъ.
[6]
Энергия диссоциации двухатомной молекулы равна энергии связи двух атомов.
[7]
Для энергии диссоциации двухатомной молекулы XY сохраняется то же определение. Продуктами процесса диссоциации являются атомы X и Y в своих основных состояниях.
[8]
Складывая энергию диссоциации двухатомных молекул с энергией их испарения из стандартных состояний ( жидкий бром, твердые иод и астат), получаем стандартные энергии атомизации простых тел.
[10]
В ряде случаев энергия диссоциации двухатомной молекулы может быть определена из анализа предиссоциации в ее спектре. Если спектр обрывается ( или размывается) в результате предиссоциации на отдельных вращательных линиях в нескольких полосах данной системы полос, экстраполяция к границе диссоциации может быть проведена с высокой точностью.
[11]
На акую величину отличается энергия диссоциации двухатомной молекулы DO определяемая экспериментально, от энергии диссоциации De, отсчитываемой от минимума кривой потенциальной энергии.
[12]
В таблице приведены значения энергий диссоциации двухатомных молекул. Расположение выдержано в алфавитном порядке.
[13]
В табл. 1 приведены значения энергий диссоциации двухатомных молекул. Расположение молекул выдержано в алфавитном порядке. Однако соединения металлов с металлоидами помещены на место соединений данного металла. В соответствии с этим CuBr идет после Gu2, а не после BrCl, также как ZnGl после ZnBr, а не после СЮ.
[14]
По сравнению с задачей расчета энергий диссоциации двухатомных молекул теоретическое исследование связи в твердых телах с помощью кванто-во-механических принципов оказывается значительно более трудным — В принципе можно написать выражение для энергии сцепления [8] через посредство атомных волновых функций, но до настоящего времени результаты, имеющие термохимическую значимость, не были получены.
[15]
Страницы:
1
2
3
4