Not to be confused with Entropy.
Enthalpy , a property of a thermodynamic system, is the sum of the system’s internal energy and the product of its pressure and volume.[1] It is a state function used in many measurements in chemical, biological, and physical systems at a constant pressure, which is conveniently provided by the large ambient atmosphere. The pressure–volume term expresses the work required to establish the system’s physical dimensions, i.e. to make room for it by displacing its surroundings.[2][3] The pressure-volume term is very small for solids and liquids at common conditions, and fairly small for gases. Therefore, enthalpy is a stand-in for energy in chemical systems; bond, lattice, solvation and other «energies» in chemistry are actually enthalpy differences. As a state function, enthalpy depends only on the final configuration of internal energy, pressure, and volume, not on the path taken to achieve it.
In the International System of Units (SI), the unit of measurement for enthalpy is the joule. Other historical conventional units still in use include the calorie and the British thermal unit (BTU).
The total enthalpy of a system cannot be measured directly because the internal energy contains components that are unknown, not easily accessible, or are not of interest in thermodynamics. In practice, a change in enthalpy is the preferred expression for measurements at constant pressure because it simplifies the description of energy transfer. When transfer of matter into or out of the system is also prevented and no electrical or shaft work is done, at constant pressure the enthalpy change equals the energy exchanged with the environment by heat.
In chemistry, the standard enthalpy of reaction is the enthalpy change when reactants in their standard states (p = 1 bar; usually T = 298 K) change to products in their standard states.[4] This quantity is the standard heat of reaction at constant pressure and temperature, but it can be measured by calorimetric methods even if the temperature does vary during the measurement, provided that the initial and final pressure and temperature correspond to the standard state. The value does not depend on the path from initial to final state because enthalpy is a state function.
Enthalpies of chemical substances are usually listed for 1 bar (100 kPa) pressure as a standard state. Enthalpies and enthalpy changes for reactions vary as a function of temperature,[5] but tables generally list the standard heats of formation of substances at 25 °C (298 K). For endothermic (heat-absorbing) processes, the change ΔH is a positive value; for exothermic (heat-releasing) processes it is negative.
The enthalpy of an ideal gas is independent of its pressure or volume, and depends only on its temperature, which correlates to its thermal energy. Real gases at common temperatures and pressures often closely approximate this behavior, which simplifies practical thermodynamic design and analysis.
Definition[edit]
The enthalpy H of a thermodynamic system is defined as the sum of its internal energy and the product of its pressure and volume:[1]
- H = U + pV,
where U is the internal energy, p is pressure, and V is the volume of the system; pV is sometimes referred to as the pressure energy ƐP.[citation needed]
Enthalpy is an extensive property; it is proportional to the size of the system (for homogeneous systems). As intensive properties, the specific enthalpy h = H/m is referenced to a unit of mass m of the system, and the molar enthalpy Hm is H/n, where n is the number of moles. For inhomogeneous systems the enthalpy is the sum of the enthalpies of the component subsystems:

where
- H is the total enthalpy of all the subsystems,
- k refers to the various subsystems,
- Hk refers to the enthalpy of each subsystem.
A closed system may lie in thermodynamic equilibrium in a static gravitational field, so that its pressure p varies continuously with altitude, while, because of the equilibrium requirement, its temperature T is invariant with altitude. (Correspondingly, the system’s gravitational potential energy density also varies with altitude.) Then the enthalpy summation becomes an integral:

where
- ρ («rho») is density (mass per unit volume),
- h is the specific enthalpy (enthalpy per unit mass),
- (ρh) represents the enthalpy density (enthalpy per unit volume),
- dV denotes an infinitesimally small element of volume within the system, for example, the volume of an infinitesimally thin horizontal layer, the integral therefore represents the sum of the enthalpies of all the elements of the volume.
The enthalpy of a closed homogeneous system is its energy function H(S,p), with its entropy S[p] and its pressure p as natural state variables which provide a differential relation for  of the simplest form, derived as follows. We start from the first law of thermodynamics for closed systems for an infinitesimal process:
of the simplest form, derived as follows. We start from the first law of thermodynamics for closed systems for an infinitesimal process:

where
- 𝛿Q is a small amount of heat added to the system,
- 𝛿W is a small amount of work performed by the system.
In a homogeneous system in which only reversible processes or pure heat transfer are considered, the second law of thermodynamics gives 𝛿Q = T dS, with T the absolute temperature and dS the infinitesimal change in entropy S of the system. Furthermore, if only pV work is done, 𝛿W = p dV. As a result,

Adding d(pV) to both sides of this expression gives

or

So

and the coefficients of the natural variable differentials  and
and  are just the single variables
are just the single variables  and
and  .
.
Other expressions[edit]
The above expression of dH in terms of entropy and pressure may be unfamiliar to some readers. There are also expressions in terms of more directly measurable variables such as temperature and pressure:[6]: 88 [7]

Here Cp is the heat capacity at constant pressure and α is the coefficient of (cubic) thermal expansion:

With this expression one can, in principle, determine the enthalpy if Cp and V are known as functions of p and T. However the expression is more complicated than  because T is not a natural variable for the enthalpy H.
because T is not a natural variable for the enthalpy H.
At constant pressure,  so that
so that  For an ideal gas, reduces to this form even if the process involves a pressure change, because αT = 1,[note 1].
For an ideal gas, reduces to this form even if the process involves a pressure change, because αT = 1,[note 1].
In a more general form, the first law describes the internal energy with additional terms involving the chemical potential and the number of particles of various types. The differential statement for dH then becomes

where μi is the chemical potential per particle for an i-type particle, and Ni is the number of such particles. The last term can also be written as μi dni (with dni the number of moles of component i added to the system and, in this case, μi the molar chemical potential) or as μi dmi (with dmi the mass of component i added to the system and, in this case, μi the specific chemical potential).
Characteristic functions and natural state variables[edit]
The enthalpy, H(S[p], p, {Ni}), expresses the thermodynamics of a system in the energy representation. As a function of state, its arguments include both one intensive and several extensive state variables. The state variables S[p], p, and {Ni} are said to be the natural state variables in this representation. They are suitable for describing processes in which they are determined by factors in the surroundings. For example, when a virtual parcel of atmospheric air moves to a different altitude, the pressure surrounding it changes, and the process is often so rapid that there is too little time for heat transfer. This is the basis of the so-called adiabatic approximation that is used in meteorology.[8]
Conjugate with the enthalpy, with these arguments, the other characteristic function of state of a thermodynamic system is its entropy, as a function, S[p](H, p, {Ni}), of the same list of variables of state, except that the entropy, S[p], is replaced in the list by the enthalpy, H. It expresses the entropy representation. The state variables H, p, and {Ni} are said to be the natural state variables in this representation. They are suitable for describing processes in which they are experimentally controlled. For example, H and p can be controlled by allowing heat transfer, and by varying only the external pressure on the piston that sets the volume of the system.[9][10][11]
Physical interpretation[edit]
The U term is the energy of the system, and the pV term can be interpreted as the work that would be required to «make room» for the system if the pressure of the environment remained constant. When a system, for example, n moles of a gas of volume V at pressure p and temperature T, is created or brought to its present state from absolute zero, energy must be supplied equal to its internal energy U plus pV, where pV is the work done in pushing against the ambient (atmospheric) pressure.
In physics and statistical mechanics it may be more interesting to study the internal properties of a constant-volume system and therefore the internal energy is used.[12][13] In chemistry, experiments are often conducted at constant atmospheric pressure, and the pressure–volume work represents a small, well-defined energy exchange with the atmosphere, so that ΔH is the appropriate expression for the heat of reaction. For a heat engine, the change in its enthalpy after a full cycle is equal to zero, since the final and initial state are equal.
Relationship to heat[edit]
In order to discuss the relation between the enthalpy increase and heat supply, we return to the first law for closed systems, with the physics sign convention: dU = δQ − δW, where the heat δQ is supplied by conduction, radiation, Joule heating. We apply it to the special case with a constant pressure at the surface. In this case the work is given by p dV (where p is the pressure at the surface, dV is the increase of the volume of the system). Cases of long range electromagnetic interaction require further state variables in their formulation, and are not considered here. In this case the first law reads:

Now,

So

If the system is under constant pressure, dp = 0 and consequently, the increase in enthalpy of the system is equal to the heat added:

This is why the now-obsolete term heat content was used in the 19th century.
Applications[edit]
In thermodynamics, one can calculate enthalpy by determining the requirements for creating a system from «nothingness»; the mechanical work required, pV, differs based upon the conditions that obtain during the creation of the thermodynamic system.
Energy must be supplied to remove particles from the surroundings to make space for the creation of the system, assuming that the pressure p remains constant; this is the pV term. The supplied energy must also provide the change in internal energy, U, which includes activation energies, ionization energies, mixing energies, vaporization energies, chemical bond energies, and so forth. Together, these constitute the change in the enthalpy U + pV. For systems at constant pressure, with no external work done other than the pV work, the change in enthalpy is the heat received by the system.
For a simple system with a constant number of particles at constant pressure, the difference in enthalpy is the maximum amount of thermal energy derivable from an isobaric thermodynamic process.[14]
Heat of reaction[edit]
The total enthalpy of a system cannot be measured directly; the enthalpy change of a system is measured instead. Enthalpy change is defined by the following equation:

where
- ΔH is the «enthalpy change»,
- Hf is the final enthalpy of the system (in a chemical reaction, the enthalpy of the products or the system at equilibrium),
- Hi is the initial enthalpy of the system (in a chemical reaction, the enthalpy of the reactants).
For an exothermic reaction at constant pressure, the system’s change in enthalpy, ΔH, is negative due to the products of the reaction having a smaller enthalpy than the reactants, and equals the heat released in the reaction if no electrical or shaft work is done. In other words, the overall decrease in enthalpy is achieved by the generation of heat.[15] Conversely, for a constant-pressure endothermic reaction, ΔH is positive and equal to the heat absorbed in the reaction.
From the definition of enthalpy as H = U + pV, the enthalpy change at constant pressure is ΔH = ΔU + p ΔV. However for most chemical reactions, the work term p ΔV is much smaller than the internal energy change ΔU, which is approximately equal to ΔH. As an example, for the combustion of carbon monoxide 2 CO(g) + O2(g) → 2 CO2(g), ΔH = −566.0 kJ and ΔU = −563.5 kJ.[16] Since the differences are so small, reaction enthalpies are often described as reaction energies and analyzed in terms of bond energies.
Specific enthalpy[edit]
The specific enthalpy of a uniform system is defined as h = H/m where m is the mass of the system. The SI unit for specific enthalpy is joule per kilogram. It can be expressed in other specific quantities by h = u + pv, where u is the specific internal energy, p is the pressure, and v is specific volume, which is equal to 1/ρ, where ρ is the density.
Enthalpy changes[edit]
An enthalpy change describes the change in enthalpy observed in the constituents of a thermodynamic system when undergoing a transformation or chemical reaction. It is the difference between the enthalpy after the process has completed, i.e. the enthalpy of the products assuming that the reaction goes to completion, and the initial enthalpy of the system, namely the reactants. These processes are specified solely by their initial and final states, so that the enthalpy change for the reverse is the negative of that for the forward process.
A common standard enthalpy change is the enthalpy of formation, which has been determined for a large number of substances. Enthalpy changes are routinely measured and compiled in chemical and physical reference works, such as the CRC Handbook of Chemistry and Physics. The following is a selection of enthalpy changes commonly recognized in thermodynamics.
When used in these recognized terms the qualifier change is usually dropped and the property is simply termed enthalpy of ‘process’. Since these properties are often used as reference values it is very common to quote them for a standardized set of environmental parameters, or standard conditions, including:
- A pressure of one atmosphere (1 atm or 101.325 kPa) or 1 bar
- A temperature of 25 °C or 298.15 K
- A concentration of 1.0 M when the element or compound is present in solution
- Elements or compounds in their normal physical states, i.e. standard state
For such standardized values the name of the enthalpy is commonly prefixed with the term standard, e.g. standard enthalpy of formation.
Chemical properties:
- Enthalpy of reaction, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of substance reacts completely.
- Enthalpy of formation, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of a compound is formed from its elementary antecedents.
- Enthalpy of combustion, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of a substance burns completely with oxygen.
- Enthalpy of hydrogenation, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of an unsaturated compound reacts completely with an excess of hydrogen to form a saturated compound.
- Enthalpy of atomization, defined as the enthalpy change required to separate one mole of a substance into its constituent atoms completely.
- Enthalpy of neutralization, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of water is formed when an acid and a base react.
- Standard Enthalpy of solution, defined as the enthalpy change observed in a constituent of a thermodynamic system when one mole of a solute is dissolved completely in an excess of solvent, so that the solution is at infinite dilution.
- Standard enthalpy of Denaturation (biochemistry), defined as the enthalpy change required to denature one mole of compound.
- Enthalpy of hydration, defined as the enthalpy change observed when one mole of gaseous ions are completely dissolved in water forming one mole of aqueous ions.
Physical properties:
- Enthalpy of fusion, defined as the enthalpy change required to completely change the state of one mole of substance from solid to liquid.
- Enthalpy of vaporization, defined as the enthalpy change required to completely change the state of one mole of substance from liquid to gas.
- Enthalpy of sublimation, defined as the enthalpy change required to completely change the state of one mole of substance from solid to gas.
- Lattice enthalpy, defined as the energy required to separate one mole of an ionic compound into separated gaseous ions to an infinite distance apart (meaning no force of attraction).
- Enthalpy of mixing, defined as the enthalpy change upon mixing of two (non-reacting) chemical substances.
Open systems[edit]
In thermodynamic open systems, mass (of substances) may flow in and out of the system boundaries. The first law of thermodynamics for open systems states: The increase in the internal energy of a system is equal to the amount of energy added to the system by mass flowing in and by heating, minus the amount lost by mass flowing out and in the form of work done by the system:

where Uin is the average internal energy entering the system, and Uout is the average internal energy leaving the system.

During steady, continuous operation, an energy balance applied to an open system equates shaft work performed by the system to heat added plus net enthalpy added
The region of space enclosed by the boundaries of the open system is usually called a control volume, and it may or may not correspond to physical walls. If we choose the shape of the control volume such that all flow in or out occurs perpendicular to its surface, then the flow of mass into the system performs work as if it were a piston of fluid pushing mass into the system, and the system performs work on the flow of mass out as if it were driving a piston of fluid. There are then two types of work performed: flow work described above, which is performed on the fluid (this is also often called pV work), and shaft work, which may be performed on some mechanical device such as a turbine or pump.
These two types of work are expressed in the equation

Substitution into the equation above for the control volume (cv) yields:

The definition of enthalpy, H, permits us to use this thermodynamic potential to account for both internal energy and pV work in fluids for open systems:

If we allow also the system boundary to move (e.g. due to moving pistons), we get a rather general form of the first law for open systems.[17] In terms of time derivatives it reads:

with sums over the various places k where heat is supplied, mass flows into the system, and boundaries are moving. The Ḣk terms represent enthalpy flows, which can be written as

with ṁk the mass flow and ṅk the molar flow at position k respectively. The term dVk/dt represents the rate of change of the system volume at position k that results in pV power done by the system. The parameter P represents all other forms of power done by the system such as shaft power, but it can also be, say, electric power produced by an electrical power plant.
Note that the previous expression holds true only if the kinetic energy flow rate is conserved between system inlet and outlet.[clarification needed] Otherwise, it has to be included in the enthalpy balance. During steady-state operation of a device (see turbine, pump, and engine), the average dU/dt may be set equal to zero. This yields a useful expression for the average power generation for these devices in the absence of chemical reactions:

where the angle brackets denote time averages. The technical importance of the enthalpy is directly related to its presence in the first law for open systems, as formulated above.
Diagrams[edit]

T–s diagram of nitrogen.[18] The red curve at the left is the melting curve. The red dome represents the two-phase region with the low-entropy side the saturated liquid and the high-entropy side the saturated gas. The black curves give the T–s relation along isobars. The pressures are indicated in bar. The blue curves are isenthalps (curves of constant enthalpy). The values are indicated in blue in kJ/kg. The specific points a, b, etc., are treated in the main text.
The enthalpy values of important substances can be obtained using commercial software. Practically all relevant material properties can be obtained either in tabular or in graphical form. There are many types of diagrams, such as h–T diagrams, which give the specific enthalpy as function of temperature for various pressures, and h–p diagrams, which give h as function of p for various T. One of the most common diagrams is the temperature–specific entropy diagram (T–s diagram). It gives the melting curve and saturated liquid and vapor values together with isobars and isenthalps. These diagrams are powerful tools in the hands of the thermal engineer.
Some basic applications[edit]
The points a through h in the figure play a role in the discussion in this section.
-
Point T (K) p (bar) s (kJ/(kg K)) h (kJ/kg) a 300 1 6.85 461 b 380 2 6.85 530 c 300 200 5.16 430 d 270 1 6.79 430 e 108 13 3.55 100 f 77.2 1 3.75 100 g 77.2 1 2.83 28 h 77.2 1 5.41 230
Points e and g are saturated liquids, and point h is a saturated gas.
Throttling[edit]
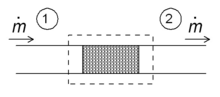
Schematic diagram of a throttling in the steady state. Fluid enters the system (dotted rectangle) at point 1 and leaves it at point 2. The mass flow is ṁ.
One of the simple applications of the concept of enthalpy is the so-called throttling process, also known as Joule–Thomson expansion. It concerns a steady adiabatic flow of a fluid through a flow resistance (valve, porous plug, or any other type of flow resistance) as shown in the figure. This process is very important, since it is at the heart of domestic refrigerators, where it is responsible for the temperature drop between ambient temperature and the interior of the refrigerator. It is also the final stage in many types of liquefiers.
For a steady state flow regime, the enthalpy of the system (dotted rectangle) has to be constant. Hence

Since the mass flow is constant, the specific enthalpies at the two sides of the flow resistance are the same:

that is, the enthalpy per unit mass does not change during the throttling. The consequences of this relation can be demonstrated using the T−s diagram above.
Example 1[edit]
Point c is at 200 bar and room temperature (300 K). A Joule–Thomson expansion from 200 bar to 1 bar follows a curve of constant enthalpy of roughly 425 kJ/kg (not shown in the diagram) lying between the 400 and 450 kJ/kg isenthalps and ends in point d, which is at a temperature of about 270 K. Hence the expansion from 200 bar to 1 bar cools nitrogen from 300 K to 270 K. In the valve, there is a lot of friction, and a lot of entropy is produced, but still the final temperature is below the starting value.
Example 2[edit]
Point e is chosen so that it is on the saturated liquid line with h = 100 kJ/kg. It corresponds roughly with p = 13 bar and T = 108 K. Throttling from this point to a pressure of 1 bar ends in the two-phase region (point f). This means that a mixture of gas and liquid leaves the throttling valve. Since the enthalpy is an extensive parameter, the enthalpy in f (hf) is equal to the enthalpy in g (hg) multiplied by the liquid fraction in f (xf) plus the enthalpy in h (hh) multiplied by the gas fraction in f (1 − xf). So

With numbers: 100 = xf × 28 + (1 − xf) × 230, so xf = 0.64. This means that the mass fraction of the liquid in the liquid–gas mixture that leaves the throttling valve is 64%.
Compressors[edit]

Schematic diagram of a compressor in the steady state. Fluid enters the system (dotted rectangle) at point 1 and leaves it at point 2. The mass flow is ṁ. A power P is applied and a heat flow Q̇ is released to the surroundings at ambient temperature Ta.
A power P is applied e.g. as electrical power. If the compression is adiabatic, the gas temperature goes up. In the reversible case it would be at constant entropy, which corresponds with a vertical line in the T–s diagram. For example, compressing nitrogen from 1 bar (point a) to 2 bar (point b) would result in a temperature increase from 300 K to 380 K. In order to let the compressed gas exit at ambient temperature Ta, heat exchange, e.g. by cooling water, is necessary. In the ideal case the compression is isothermal. The average heat flow to the surroundings is Q̇. Since the system is in the steady state the first law gives

The minimal power needed for the compression is realized if the compression is reversible. In that case the second law of thermodynamics for open systems gives

Eliminating Q̇ gives for the minimal power

For example, compressing 1 kg of nitrogen from 1 bar to 200 bar costs at least (hc − ha) − Ta(sc − sa). With the data, obtained with the T–s diagram, we find a value of (430 − 461) − 300 × (5.16 − 6.85) = 476 kJ/kg.
The relation for the power can be further simplified by writing it as

With dh = T ds + v dp, this results in the final relation

History and etymology[edit]
The term enthalpy was coined relatively late in the history of thermodynamics, in the early 20th century. Energy was introduced in a modern sense by Thomas Young in 1802, while entropy was coined by Rudolf Clausius in 1865. Energy uses the root of the Greek word ἔργον (ergon), meaning «work», to express the idea of capacity to perform work. Entropy uses the Greek word τροπή (tropē) meaning transformation or turning. Enthalpy uses the root of the Greek word θάλπος (thalpos) «warmth, heat».[19]
The term expresses the obsolete concept of heat content,[20] as dH refers to the amount of heat gained in a process at constant pressure only,[21] but not in the general case when pressure is variable.[22]
Josiah Willard Gibbs used the term «a heat function for constant pressure» for clarity.[note 2]
Introduction of the concept of «heat content» H is associated with Benoît Paul Émile Clapeyron and Rudolf Clausius (Clausius–Clapeyron relation, 1850).
The term enthalpy first appeared in print in 1909.[23] It is attributed to Heike Kamerlingh Onnes, who most likely introduced it orally the year before, at the first meeting of the Institute of Refrigeration in Paris.[24]
It gained currency only in the 1920s, notably with the Mollier Steam Tables and Diagrams, published in 1927.
Until the 1920s, the symbol H was used, somewhat inconsistently, for «heat» in general.
The definition of H as strictly limited to enthalpy or «heat content at constant pressure» was formally proposed by Alfred W. Porter in 1922.[25][26]
See also[edit]
- Standard enthalpy of formation
- Calorimetry
- Calorimeter
- Departure function
- Hess’s law
- Isenthalpic process
- Laws of thermodynamics
- Stagnation enthalpy
- Thermodynamic databases for pure substances
Notes[edit]
- ^

- ^ The Collected Works of J. Willard Gibbs, Vol. I do not contain reference to the word enthalpy, but rather reference the «heat function for constant pressure». See: Henderson, Douglas; Eyring, Henry; Jost, Wilhelm (1967). Physical Chemistry: An Advanced Treatise. Academic Press. p. 29.

References[edit]
- ^ a b IUPAC, Compendium of Chemical Terminology, 2nd ed. (the «Gold Book») (1997). Online corrected version: (2006–) «enthalpy». doi:10.1351/goldbook.E02141
- ^ Zemansky, Mark W. (1968). «Chapter 11». Heat and Thermodynamics (5th ed.). New York, NY: McGraw-Hill. p. 275.
- ^ Van Wylen, G. J.; Sonntag, R. E. (1985). «Section 5.5». Fundamentals of Classical Thermodynamics (3rd ed.). New York: John Wiley & Sons. ISBN 978-0-471-82933-1.
- ^ Atkins, Peter; de Paula, Julio (2006). Atkins’ Physical Chemistry (8th ed.). W.H.Freeman. p. 51. ISBN 0-7167-8759-8.
- ^ Laidler, Keith J.; Meiser, John H. (1999). Physical Chemistry (3 ed.). Boston: Houghton Mifflin. p. 66. ISBN 0-395-91848-0.
- ^ Guggenheim, E. A. (1959). Thermodynamics. Amsterdam: North-Holland Publishing Company.
- ^ Moran, M. J.; Shapiro, H. N. (2006). Fundamentals of Engineering Thermodynamics (5th ed.). John Wiley & Sons. p. 511. ISBN 9780470030370.
- ^ Iribarne, J.V., Godson, W.L. (1981). Atmospheric Thermodynamics, 2nd edition, Kluwer Academic Publishers, Dordrecht, ISBN 90-277-1297-2, pp. 235–236.
- ^ Tschoegl, N.W. (2000). Fundamentals of Equilibrium and Steady-State Thermodynamics, Elsevier, Amsterdam, ISBN 0-444-50426-5, p. 17.
- ^ Callen, H. B. (1960/1985), Thermodynamics and an Introduction to Thermostatistics, (first edition 1960), second edition 1985, John Wiley & Sons, New York, ISBN 0-471-86256-8, Chapter 5.
- ^ Münster, A. (1970), Classical Thermodynamics, translated by E. S. Halberstadt, Wiley–Interscience, London, ISBN 0-471-62430-6, p. 6.
- ^ Reif, F. (1967). Statistical Physics. London: McGraw-Hill.
- ^ Kittel, C.; Kroemer, H. (1980). Thermal Physics. London: Freeman.
- ^ Rathakrishnan (2015). High Enthalpy Gas Dynamics. John Wiley and Sons Singapore Pte. Ltd. ISBN 978-1118821893.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry. Benjamin/Cummings. p. 53. ISBN 978-0-8053-5682-3.
- ^ Petrucci, Ralph H.; Harwood, William S.; Herring, F. Geoffrey (2002). General Chemistry (8th ed.). Prentice Hall. pp. 237–238. ISBN 978-0-13-014329-7.
- ^ Moran, M. J.; Shapiro, H. N. (2006). Fundamentals of Engineering Thermodynamics (5th ed.). John Wiley & Sons. p. 129. ISBN 9780470030370.
- ^ Figure composed with data obtained with RefProp, NIST Standard Reference Database 23.
- ^ θάλπος in A Greek–English Lexicon.
- ^ Howard (2002) quotes J. R. Partington in An Advanced Treatise on Physical Chemistry (1949) as saying that the function H was «usually called the heat content».
- ^ Tinoco, Ignacio Jr.; Sauer, Kenneth; Wang, James C. (1995). Physical Chemistry (3rd ed.). Prentice-Hall. p. 41. ISBN 978-0-13-186545-7.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry. Benjamin/Cummings. p. 53. ISBN 978-0-8053-5682-3.
- ^ Dalton, J. P. (1909). «Researches on the Joule–Kelvin-effect, especially at low temperatures. I. Calculations for hydrogen». Proceedings of the Section of Sciences (Koninklijke Akademie van Wetenschappen te Amsterdam [Royal Academy of Sciences at Amsterdam]). 11 (part 2): 863–873. Bibcode:1908KNAB…11..863D. ; see p. 864, footnote (1).
- ^ See:
- Laidler, Keith (1995). The World of Physical Chemistry. Oxford University Press. p. 110.
- Van Ness, Hendrick C. (2003). «H Is for Enthalpy». Journal of Chemical Education. 80 (6): 486. Bibcode:2003JChEd..80..486V. doi:10.1021/ed080p486.1.
- ^ Porter, Alfred W. (1922). «The generation and utilisation of cold. A general discussion». Transactions of the Faraday Society. 18: 139–143. doi:10.1039/tf9221800139.; see p. 140.
- ^
Howard, Irmgard (2002). «H Is for Enthalpy, Thanks to Heike Kamerlingh Onnes and Alfred W. Porter». Journal of Chemical Education. 79 (6): 697. Bibcode:2002JChEd..79..697H. doi:10.1021/ed079p697.
Bibliography[edit]
- Dalton, J.P. (1909). «Researches on the Joule–Kelvin effect, especially at low temperatures. I. Calculations for hydrogen» (PDF). KNAW Proceedings. 11: 863–873. Bibcode:1908KNAB…11..863D.
- Haase, R. (1971). Jost, W. (ed.). Physical Chemistry: An Advanced Treatise. New York: Academic. p. 29.
- Gibbs, J. W. The Collected Works of J. Willard Gibbs, Vol. I (1948 ed.). New Haven, CT: Yale University Press. p. 88.
- Howard, I. K. (2002). «H Is for Enthalpy, Thanks to Heike Kamerlingh Onnes and Alfred W. Porter». J. Chem. Educ. 79 (6): 697–698. Bibcode:2002JChEd..79..697H. doi:10.1021/ed079p697.
- Laidler, K. (1995). The World of Physical Chemistry. Oxford: Oxford University Press. p. 110. ISBN 978-0-19-855597-1.
- Kittel, C.; Kroemer, H. (1980). Thermal Physics. New York: S. R. Furphy & Co. p. 246.
- DeHoff, R. (2006). Thermodynamics in Materials Science. CRC Press. ISBN 9780849340659.
External links[edit]
- Enthalpy – Eric Weisstein’s World of Physics
- Enthalpy – Georgia State University
- Enthalpy example calculations – Texas A&M University Chemistry Department
![]()
Загрузить PDF
![]()
Загрузить PDF
Во время химических реакций происходит поглощение или выделение тепла в окружающую среду. Такой теплообмен между химической реакцией и окружающей средой называется энтальпией, или H. Однако измерить энтальпию напрямую невозможно, поэтому принято рассчитывать изменение температуры окружающей среды (обозначаемое ∆H). ∆H показывает, что в ходе химической реакции происходит выделение тепла в окружающую среду (экзотермическая реакция) или поглощение тепла (эндотермическая реакция). Рассчитывается энтальпия так: ∆H = m x s x ∆T, где m — масса реагентов, s — теплоемкость продукта реакции, ∆T — изменение температуры в результате реакции.
-
1
Определите реагенты и продукты реакции. Любая химическая реакция имеет реагенты и продукты реакции. Продукт реакции создается в результате взаимодействия реагентов. Другими словами реагенты — это ингредиенты в рецепте, а продукт реакции — это готовое блюдо. Чтобы найти ∆H реакции, необходимо знать реагенты и продукты реакции.
- Например, необходимо найти энтальпию реакции образования воды из водорода и кислорода: 2H2 (водород) + O2 (кислород) → 2H2O (вода). В этой реакции H2 и O2 – реагенты, а H2O — продукт реакции.
-
2
Определите общую массу реагентов. Далее необходимо подсчитать массу реагентов. Если вы не можете взвесить их, то подсчитайте молекулярную массу, чтобы найти фактическую. Молекулярная масса — это постоянная, которую можно найти в периодической таблице Менделеева или в других таблицах молекул и соединений. Умножьте массу каждого реагента на число молей.
- В нашем примере реагенты водород и кислород имеют молекулярные массы 2 г и 32 г соответственно. Поскольку мы используем 2 моль водорода (коэффициент в химической реакции перед водородом H2) и 1 моль кислорода (отсутствие коэффициента перед O2 обозначает 1 моль), то общая масса реагентов рассчитывается следующим образом:
2 × (2 г) + 1 × (32 г) = 4 г + 32 г = 36 г
- В нашем примере реагенты водород и кислород имеют молекулярные массы 2 г и 32 г соответственно. Поскольку мы используем 2 моль водорода (коэффициент в химической реакции перед водородом H2) и 1 моль кислорода (отсутствие коэффициента перед O2 обозначает 1 моль), то общая масса реагентов рассчитывается следующим образом:
-
3
Определите теплоемкость продукта. Далее определите теплоемкость продукта реакции. Каждая молекула имеет определенную величину теплоемкости, которая является постоянной. Найдите эту постоянную в таблицах учебника по химии. Существует несколько единиц измерения теплоемкости; в наших расчетах мы будем использовать Дж/г°C.
- Обратите внимание на то, что при наличии нескольких продуктов реакции вам потребуется рассчитать теплоемкость каждого, а затем сложить их, чтоб получить энтальпию всей реакции.
- В нашем примере, продукт реакции — вода, которая имеет теплоемкость 4,2 Дж/г°C.
-
4
Найдите изменение температуры. Теперь мы найдем ∆T — разницу температур до и после реакции. Из начальной температуры (T1) вычтите конечную температуру (T2). Чаще всего в задачах по химии используется шкала Кельвина (К) (хотя по шкале Цельсия (°С) получится тот же результат).
- В нашем примере давайте предположим, что начальная температура реакции была 185 K, а после реакции стала 95 K, значит, ∆T вычисляется так:
∆T = T2 – T1 = 95 K — 185 K = -90 K
- В нашем примере давайте предположим, что начальная температура реакции была 185 K, а после реакции стала 95 K, значит, ∆T вычисляется так:
-
5
Найдите энтальпию по формуле ∆H = m x s x ∆T. Если известна m — масса реагентов, s — теплоемкость продукта реакции и ∆T — изменение температуры, то можно подсчитать энтальпию реакции. Подставьте значения в формулу ∆H = m x s x ∆T и получите энтальпию. Результат вычисляется в Джоулях (Дж).
- В нашем примере энтальпия вычисляется так:
∆H = (36 г) × (4,2 ДжK — 1 г — 1) × (-90 K) = -13608 Дж
- В нашем примере энтальпия вычисляется так:
-
6
Определите, выделяется или поглощается энергия в ходе рассматриваемой реакции. Одна из самых распространенных причин, по которой требуется вычислить ∆H на практике, — узнать, будет ли реакция экзотермической (выделение тепла и снижение собственной энергии) или эндотермической (поглощение тепла из окружающей среды и повышение собственной энергии). Если значение ∆H положительное, значит, реакция эндотермическая. Если отрицательное, значит, реакция экзотермическая. Чем больше абсолютное значение ∆H, тем больше энергии выделяется или поглощается. Будьте осторожны, если собираетесь проводить практический опыт: во время реакций с высоким значением энтальпии может произойти большое высвобождение энергии, и если оно протекает быстро, то может привести ко взрыву.
- В нашем примере конечный результат получился равным -13608 Дж. Перед значением энтальпии отрицательный знак, а это означает, что реакция экзотермическая. Горячие газы (в виде пара) H2 и O2 должны выделить некоторое количество тепла, чтобы образовать молекулу воды, то есть реакция образования H2O является экзотермической.
Реклама






-
1
Подсчитайте энергию связей для оценки энтальпии. Почти все химические реакции приводят к разрыву одних связей и образованию других. Энергия в результате реакции не возникает ниоткуда и не разрушается: это та энергия, которая требуется для разрыва или образования этих связей. Поэтому изменение энтальпии всей реакции можно довольно точно оценить путем суммирования энергии этих связей.
- Например, рассмотрим реакцию: H2 + F2 → 2HF. В этом случае, энергия для разрыва связи в молекуле H2 составляет 436 кДж/моль, а энергия для разрыва F2 составляет 158 кДж/моль.[1]
Наконец, энергия необходимая для образования связи в молекуле HF из H и F = -568 кДж/моль.[2]
Умножаем последнее значение на 2, так как в ходе реакции получаем 2 моль HF: 2 × -568 = -1136 кДж/моль. Складываем значения:
436 + 158 + (-1136) = -542 кДж/моль.
- Например, рассмотрим реакцию: H2 + F2 → 2HF. В этом случае, энергия для разрыва связи в молекуле H2 составляет 436 кДж/моль, а энергия для разрыва F2 составляет 158 кДж/моль.[1]
-
2
Используйте энтальпию образования для оценки энтальпии. Энтальпия образования позволяет рассчитать ∆H через вычисление реакций образования реагентов и продуктов. Если известна энтальпия образования продуктов реакции и реагентов, то вы можете оценить энтальпию в целом путем сложения, как и в случае энергии, рассмотренном выше.
- Например, рассмотрим следующую реакцию: C2H5OH + 3O2 → 2CO2 + 3H2O. Мы знаем, что энтальпия образования рассчитывается:[3]
C2H5OH → 2C + 3H2 + 0,5O2 = 228 кДж/моль
2C + 2O2 → 2CO2 = -394 × 2 = -788 кДж/моль
3H2 + 1.5 O2 → 3H2O = -286 × 3 = -858 кДж/моль
Теперь необходимо сложить значения образованных веществ, полученные выше, для определения энтальпии реакции: C2H5OH + 3O2 → 2CO2 + 3H2O,
228 + -788 + -858 = -1418 кДж/моль.
- Например, рассмотрим следующую реакцию: C2H5OH + 3O2 → 2CO2 + 3H2O. Мы знаем, что энтальпия образования рассчитывается:[3]
-
3
Не забывайте о знаках перед значениями энтальпии. При вычислении энтальпии образования формулу для определения энтальпии реакции продукта вы переворачиваете, и знак энтальпии должен поменяться. Другими словами, если вы переворачиваете формулу, то знак энтальпии должен смениться на противоположный.
- В примере обратите внимание на то, что реакция образования для продукта C2H5OH записана наоборот. C2H5OH → 2C + 3H2 + 0,5O2 то есть C2H5OH распадается, а не синтезируется. Поэтому знак перед энтальпией в такой реакции положительный, 228 кДж/моль, хотя энтальпия образования C2H5OH составляет -228 кДж/моль.
Реклама



-
1
Возьмите чистую емкость и налейте туда воды. Увидеть принципы энтальпии в действии нетрудно — достаточно провести простой опыт. Необходимо, чтобы на результат эксперимента не повлияли посторонние загрязнители, так что емкость нужно вымыть и простерилизовать. Ученые для измерения энтальпии используют специальный закрытые контейнеры — калориметры, но вам вполне подойдет стеклянный стакан или колба. Заполните емкость чистой водопроводной водой комнатной температуры. Желательно проводить эксперимент в прохладном помещении.
- Для эксперимента желательно использовать небольшую емкость. Мы будем рассматривать энтальпию реакции воды с «Алка-Зельтцер», поэтому, чем меньше воды используется, тем более очевидным будет изменение температуры.
-
2
Поместите термометр в емкость. Возьмите термометр и опустите его в емкость с водой так, чтобы граница прочтения температуры была ниже уровня воды. Снимите показания термометра — это будет начальная температура, T1.
- Предположим, что температуры воды 10 °C. Мы будем использовать это значение для демонстрации принципов энтальпии.
-
3
Добавьте в емкость одну таблетку «Алка-Зельтцер». Готовы начать опыт? Бросьте в воду одну таблетку «Алка-Зельтцер». Она сразу начнет пузыриться и шипеть. Это происходит из-за реакции между бикарбонатом (HCO3—) и лимонной кислотой (H+). В результате образуются вода и углекислый газ по формуле: 3HCO3− + 3H+ → 3H2O + 3CO2.
-
4
Измерьте конечную температуру. Наблюдайте за ходом реакции: таблетка «Алка-Зельтцер» будет постепенно растворяться. Когда она растворится полностью, измерьте температуру еще раз. Вода должна стать немного холоднее. Если температура воды напротив стала выше начальной, значит, эксперименту помешали какие-то внешние факторы (например, слишком теплое помещение, где проводился эксперимент).
- Предположим, что температура составляет теперь 8 °C.
-
5
Подсчитаем энтальпию реакции. Когда таблетка «Алка-Зельтцер» вступает в реакцию с водой, образуются вода и углекислый газ (те самые шипучие пузырьки) и происходит снижение температуры (это тот результат, который должен получиться, если опыт прошел успешно). Можно сделать вывод, что данная химическая реакция является эндотермической, то есть она сопровождается поглощением энергии из окружающей среды — в данном случае из воды. В результате температура воды снижается.
- В нашем эксперименте температура воды снизилась на два градуса. Это согласуется с теорией: реакция растворения «Алка-Зельтцер» в воде эндотермическая и сопровождается небольшим поглощением энергии.
Реклама





Советы
- В подсчетах используется шкала Кельвина (K) — это температурная шкала, аналогическая шкале Цельсия, и часто применяемая в химии и физике. Чтобы перевести значение градусов Цельсия в кельвины, необходимо добавить или вычесть 273 градуса: K = °C + 273.
Реклама
Об этой статье
Эту страницу просматривали 115 967 раз.
Была ли эта статья полезной?
В
промышленности химические процессы
довольно часто проводят при повышенных
давлениях, т.е. в условиях, существенно
отличающихся от стандартных.
В разделе 2.10. уже отмечалось, что в
порядке первого приближения
влиянием давления на энтальпию
конденсированных фаз можно пренебречь.
В случае газов это влияние обнаруживается
вблизи критической области при давлениях
> ркр/2.
Эти положения справедливы не только
для индивидуальных
веществ, но и для химических реакций.
Если
химические превращения происходят с
участием газов при высоких степенях
сжатия последних, то влияние давления
на энтальпию реакции учитывается
с помощью уравнения:
![]()
/
-, ,,
где
ДГУ
— изменение объема в ходе реакции;-
температурная
![]()
•
зависимость
ДГУЖ)
Чтобы
установить в количественной форме, как
именно энтальпия процесса
изменяется с давлением, необходимо
располагать данными об уравнении
состояния газовой смеси. Но прежде, чем
приступить к вычислениям,
учитывающим влияние давления, следует
принять во внимание, что
масштабы воздействия давления на
тепловой эффект реакции будут сравнительно
невелики. Например, в случае реакции
синтеза аммиака (Arv
= -l)s
которая, как известно, осуществляется
при 400-500 атм, учёт
влияния давления
корректирует величину теплового эффекта
всего на 10%.
Вывод
этого уравнения и его анализ будет дан
в разделе 3.2.8 [p# 66]
2. Энергетика
[p# 67]
3. Критерии направленности процессов и равновесия в системах постоянного и переменного составов
3.1. Второе начало термодинамики (постулат о существовании энтропии)
Понятие
и термин «энтропия»
ввел
Р. Клаузиус в 1865 г. Слово это греческого
происхождения: эн
— предлог
«в», trope
— «превращение».
Буквально —«превращение
в». Имелось
в виду рассеяние энергии, т.е. превращение
энергии в менее ценные формы.
Первоначально
вывод о существовании данной
термодинамической функции
был сделан в результате изучения работы
тепловых машин. Со временем,
однако, было установлено, что содержание
понятия энтропия является
гораздо более емким. Оно, как оказалось,
может использоваться в качестве
критерия направленности процессов и
равновесия в изолированных системах.
Закон
сохранения энергии (в форме первого
начала термодинамики) говорит
об эквивалентности различных форм
энергии. Согласно ему возможны любые
процессы, в которых вместо одного вида
энергии появляется эквивалентное
количество энергии другого вида.
Мировой
океан, например, является гигантским
резервуаром теплоты. Спросим
себя, можно ли использовать запасенную
в нем энергию, например, для
обогрева нашего жилища? Согласно первому
закону — вполне: С этой целью необходимо
отобрать ничтожную часть теплоты у
мирового океана. В действительности,
однако, это невозможно вследствие
запрета, наложенного вторым
началом термодинамики, утверждающим,
что теплота не переходит самопроизвольно
от тела, более холодного, к телу, более
нагретому.
Известно
более двадцати определений второго
начала термодинамики. Они
были предложены многими корифеями
мировой науки — Р. Клаузиусом, У.
Гиббсом, А, Эйнштейном, М. Планком, Э.
Ферми и др. Написаны специальные
монографии, авторы которых анализируют
достоинства и недостатки
той или иной формулировки (см., например:
Путилов. К.А. Термодинамика.
М.: Наука,
1971; Кемпбел.
Дж. Современная общая химия. М.: Мир,
1975: Т. 3. С. 417).
Одни
из этих определений выражены в строгой
форме, другие имеют качественный
характер, а некоторые могут показаться
абстрактными. Например:
62
[p# 68]
• Энергия
Вселенной постоянна, а ее
энтропия стремится к
максимуму
(Р.
Клаузиус);
9
Энтропия
— это стрелка, отмеряющая время (Эдингтон)’,
-
Для
равновесия любой изолированной
системы необходимо и
достаточно,
чтобы во всех возможных изменениях
состояния системы, при
которых
не изменяется ее энергия, изменение ее
энтропии было бы нулевым
или
отрицательным (У.
Гиббс) -
Теплота
не может сама по себе переходить от
холодного к более
теплому
телу. (Р.
Клаузиус),
Последнее
из приведенных определений воспринимается
как переложение
вполне очевидного факта, что «вода
не перетекает сама по себе из низины
на возвышенность, хотя обратный процесс
происходит самопроизвольно».
Заметим, кстати, что, основываясь на
этой идее. Сади Карно*)
рассмотрел тепловую машину как водяную
мельницу, представив разность
температур как разность уровней, и
получил, опираясь на неточную аналогию,
правильное выражение для КПД идеальной
тепловой машины. Последний,
как известно, не зависит от природы
рабочего тела и вычисляется из
соотношения;
![]()
j
где
Tt
иТ2
— температуры нагревателя и холодильника,
соответственно, Q1
nQ2
-теплоты,
соответственно, взятая от нагревателя
и переданная холодильнику.
Существует
несколько определений понятия энтропии.
Все они как и второе
начало термодинамики имеют постулативный
характер и каждый из известных
подходов может рассматриваться в
качестве формулировки второго начала.
В данном курсе предпочтение будет отдано
двум определениям.
Первое из них
утверждает, что
«существует
некоторая экстенсивная функция состояния,
называемая энтропией,
изменение которой связано с поглощаемой
теплотой и температурой
системы уравнением
![]()
*)
Сади — редкое для европейца имя. Отец
Карно был
известным французским
генералом-республиканцем
и большим почитателем таланта персидского
поэта — Саади, в
честь
которого он и назвал своего сына.
[p# 69]
3.
Критерии (энтропия) . 63
[p# 70]
где
знак равенства в этом выражении относится
к равновесным (обратимым),
а знак неравенства — к неравновесным
(необратимым) процессам
«.
Второе
определение связано со статистическим
толкованием энтропии. Согласно
ему :
«изолированная
система изменяется в своем развитии от
состояний, термодинамически
менее вероятных, к состояниям, более
вероятным, или иначе,
от состояний с малой энтропией к
состояниям с большей энтропией».
Для
иллюстрации справедливости этого
положения вспомним, например,
что беспорядок в комнате всегда возникает
самопроизвольно.
Как рассчитать энтальпию
В любом веществе содержится некоторое количество тепла. Это тепло называют энтальпией. Энтальпия есть величина, характеризующая энергию системы. В физике и химии она показывает теплоту реакции. Она является альтернативой внутренней энергии, и эту величину чаще всего указывают при постоянном давлении, когда система имеет некоторый запас энергии.
Инструкция
В физико-химических процессах происходит передача тепла от одного тела к другому. Это возможно, как правило, при постоянном давлении и температуре. В роли постоянного давления обычно выступает атмосферное. Энтальпия, как и внутренняя энергия, является функцией состояния.Внутренняя энергия представляет собой сумму кинетической и потенциальной энергий всей системы. Она является основой для уравнения энтальпии. Энтальпия представляет собой сумму внутренней энергии и давления, умноженного на объем системы, и равна:H=U+pV, где p — давление в системе, V — объем системы.Вышеуказанная формула применяется для расчета энтальпии в том случае, когда даны все три величины: давление, объем и внутренняя энергия. Однако, далеко не всегда энтальпия рассчитывается таким образом. Помимо него, существует еще несколько способов вычисления энтальпии.
Зная свободную энергию и энтропию, можно вычислить энтальпию. Свободная энергия, или энергия Гиббса, представляет собой часть энтальпии системы, затраченную на превращение в работу, и равна разности энтальпии и температуры, умноженной на энтропию:ΔG=ΔH-TΔS (ΔH, ΔG, ΔS — приращения величин)Энтропия в данной формуле является мерой неупорядоченности частиц системы. Она возрастает при увеличении температуры T и давления. При ΔG<0 процесс идет самопроизвольно, при ΔG>0 — не идет.
Кроме того, энтальпия также рассчитывается исходя из уравнения химической реакции. Если дано уравнение химической реакции вида A+B=C, то энтальпию можно определить по формуле:dH=dU+ΔnRT, где Δn=nk-nн (nk и nн — число молей продуктов реакции и исходных веществ)При изобарном процессе энтропия равна изменению теплоты в системе: dq=dH.При постоянном давлении энтальпия равна:H=∫СpdTВ случае, если энтальпийный и энтропийный факторы уравновешивают друг друга, приращение энтальпии равно произведению температуры на приращение энтропии:ΔH=TΔS
Источники:
- как вычислить изменение энтропии в реакции
Войти на сайт
или
Забыли пароль?
Еще не зарегистрированы?
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
Уравнение энтальпии с использованием давления и плотности Калькулятор
| Search | ||
| Дом | Инженерное дело ↺ | |
| Инженерное дело | Механический ↺ | |
| Механический | механика жидкости ↺ | |
| механика жидкости | Гиперзвуковой поток ↺ | |
| Гиперзвуковой поток | Метод конечных разностей марша по пространству: дополнительные решения уравнений Эйлера ↺ |
|
✖Коэффициент удельной теплоемкости газа – это отношение удельной теплоемкости газа при постоянном давлении к его удельной теплоемкости при постоянном объеме.ⓘ Коэффициент удельной теплоемкости [k] |
+10% -10% |
||
|
✖Давление — это сила, приложенная перпендикулярно поверхности объекта на единицу площади, по которой распределяется эта сила.ⓘ Давление [P] |
+10% -10% |
||
|
✖Плотность материала показывает плотность этого материала в определенной заданной области. Это берется как масса на единицу объема данного объекта.ⓘ Плотность [ρ] |
+10% -10% |
|
✖Энтальпия – это термодинамическая величина, эквивалентная общему содержанию тепла в системе.ⓘ Уравнение энтальпии с использованием давления и плотности [H] |
⎘ копия |
Уравнение энтальпии с использованием давления и плотности Решение
ШАГ 0: Сводка предварительного расчета
ШАГ 1. Преобразование входов в базовый блок
Коэффициент удельной теплоемкости: 1.6 —> Конверсия не требуется
Давление: 800 паскаль —> 800 паскаль Конверсия не требуется
Плотность: 997 Килограмм на кубический метр —> 997 Килограмм на кубический метр Конверсия не требуется
ШАГ 2: Оцените формулу
ШАГ 3: Преобразуйте результат в единицу вывода
2.13975259110665 Джоуль —>0.00213975259110665 килоджоуль (Проверьте преобразование здесь)
11 Метод конечных разностей марша по пространству: дополнительные решения уравнений Эйлера Калькуляторы
Уравнение энтальпии с использованием давления и плотности формула
Энтальпия = (Коэффициент удельной теплоемкости/(Коэффициент удельной теплоемкости-1))*(Давление/Плотность)
H = (k/(k-1))*(P/ρ)
что такое энтальпия?
Энтальпия — это свойство термодинамической системы, определяемое как сумма внутренней энергии системы и произведения ее давления и объема.