Энтропия активации
Энтропия активации ΔS≠представляет собой разность между
значениями энтропии исходных соединений
и переходного состояния данной реакции.
Эта разность существенно зависит от
ограниченности или степени свободы
вращательного и поступательного
движений, а также может определяться
различиями в сольватации. Если переходное
состояние более «компактно», чем
исходная система, то в этом случае будет
существовать относительная ограниченность
в свободном движении отдельных групп
атомов; этому будет соответствовать
отрицательное значение ΔS≠.
При более свободном расположении
атомов в переходном состоянии ΔS≠будет положительным. Отсюда видно, что
как из знака, так и из абсолютного
значения ΔS≠можно
получить сведения о переходном состоянии
реакции.
Для неполярных реакций эффекты
сольватации не могут играть значительной
роли, поэтому разность энтропии в ходе
реакции по координате реакции будет
определяться только степенью свободы
вращательного и поступательного
движений. Во многих случаях определенные
значения ΔS≠типичны
для определенного механизма реакции.
Следующий пример поясняет этот тезис.
При бимолекулярной реакции две
молекулы со свободным поступательным
движением объединяются в переходном
состоянии, движущемся в растворе как
единое целое. Вследствие этого общая
энтропия уменьшается, так что при
достижении переходного состояния в
случае бимолекулярного процесса значение
ΔS≠составляет
около —20 кал/(град∙моль).
Для протекающих по мономолекулярной
схеме реакций изомеризации в
общем случае относительное расположение
отдельных групп атомов в переходном
состоянии фиксировано сильнее, чем
в исходном
веществе и в продукте.
Энтропия подобных реакций также проходит
через минимум, поэтому ΔS≠здесь также имеет отрицательное значение.
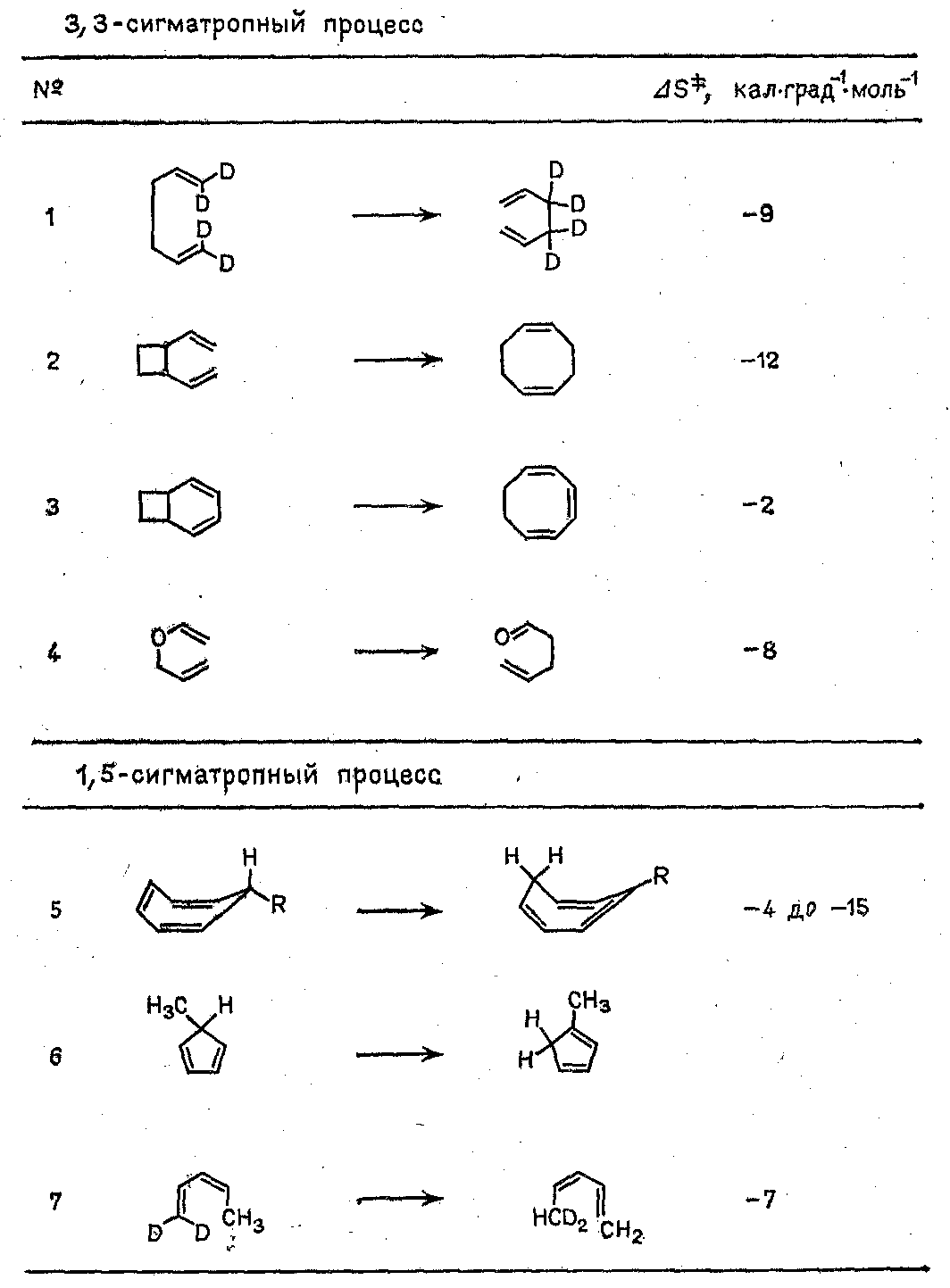
Примечательным является различие
энтропии активации при изомеризации
цис-1,2-дивинилциклобута-на и
бицикло[4.2.0]октадиена-2,4 (табл. 1;
№№ 2 и 3). В бициклической системе
свободное движение обеих винильных
групп ограничено уже в исходном соединении
из-за включения в цикл, так что при
достижении переходного состояния
энтропия уменьшается лишь незначительно.
В случае же моноциклического субстрата
обе винильные группы должны быть
фиксированы только в переходном
состоянии.
Если, же степень свободы движения
отдельных групп атомов при достижении
переходного состояния несколько
увеличивается из-за уменьшения напряжения
цикла, то ΔS≠становится положительным, хотя и
небольшим по абсолютной величине, как
в случае валентной изомеризации
бульвалена или при других
электроциклических реакциях раскрытия
цикла.
Таблица 1.
Энтропия активации
реакций валентной изомеризации
С помощью характеристических значений
энтропии активации для различных типов
реакций можно определить, проходит
ли термическое разложение этилового
эфира хлоругольной кислоты до
диоксида углерода и хлористого этила
через циклическое переходное состояние
3 или же протекает радикальный распад.

В переходном состоянии 3 отдельные
группы атомов прочно фиксированы в
пространстве, что предполагает
отрицательное значение ΔS≠.
Радикальный распад начинается с
предварительной диссоциации; исходя
из этого,ΔS≠
должно быть положительным и в пределах
от +5 до +20 кал∙град-1∙моль-1.
Экспериментально найденное значениеΔS≠составляет —
18 кал∙град-1∙моль-1, что не
согласуется с радикальным механизмом
распада и дает ясное подтверждение
внутримолекулярному протеканию реакции
через переходное состояние 3.
Для полярных реакций возникновение
и исчезновение зарядов вносит заметные
дополнительные изменения в энтропию
при перемещении вдоль координаты
реакции. В случае, когда исходная система
малополярна, окружающие молекулы
растворителя практически не ограничены
в своем движении. При возникновении
в ходе реакции зарядов переходное
состояние обладает большим дипольным
моментом, чем исходная система.
Возникающий диполь своим электрическим
полем вызывает упорядочение среди
окружающих его молекул растворителя,
что приводит к ограничению
поступательного движения. Таким образом,
увеличение сольватации переходного
состояния по сравнению с исходной
системой приводит к дополнительному
уменьшению энтропии, например:


Приведенные данные позволяют считать,
что появление в переходном состоянии
реакции пары зарядов снижает ΔS≠примернона 10-30 кал∙град-1∙моль-1,
тогда как на частьΔS≠,
обусловленную бимолекулярным
характером реакции, приходится около
-20 кал∙град-1∙моль-1. Бросается
в глаза, что изменения в энтропии,
обусловленные зарядами, больше в
неполярном растворителе (бензоле), чем
в полярном (этаноле). Это связано с
тем, что этанол как полярный растворитель
даже в отсутствие растворенных диполей
характеризуется высокой степенью
упорядоченности. Увеличение степени
упорядоченности вокруг заряженной
молекулы в этом растворителе не может
быть большим, чем в неполярном растворителе,
где в
отсутствие электрического
поля упорядоченность практически
отсутствует.
Стерические взаимодействия в
переходном состоянии реакции, как и
следовало ожидать, сказываются на
энтропии активации. Для реакцииSN2
у первичного атома углерода принимают
переходное состояние структуры 4. Здесь
углы между связями, идущими к заместителям
у атакуемого атома углерода, меньше,
чем в исходном веществе или в продукте.
Группы атомов в переходном состоянии
располагаются теснее, чем в исходной
системе, и следствием этого является
ограничение вращения этих групп в
переходном состоянии. Это должно
становиться более заметным при переходе
к более объемистым остаткамR.
Для приведенной ниже реакции энтропия
активации отрицательна, т. е.
скоростьопределяющая стадия является
бимолекулярной*.
Подводя итог, можно констатировать, что
отрицательное значение энтропии
активации свойственно реакциям
тогда, когда переходное состояние более
упорядочено и компактно по сравнению
с исходной системой. Это увеличение
порядка отвечает сумме свободной
энтальпии, которая дополнительно
привносится за счет энтальпии активации.
Напротив, если переходное состояние
менее упорядочено и занимает больший
объем по сравнению с исходным
состоянием, то более вероятно увеличение
степеней свободы вращательного и
поступательного движений. Отсюда
вытекает положительное значение
энтропии активации, так что соответствующий
выигрыш в свободной энтальпии идет на
пользу реакции, поскольку в этом
случае не должен использоваться весь
объем энтальпии активации, как следует
из уравнения Гиббса.
ΔG≠ = ΔH≠
— T ΔS≠
Соседние файлы в папке Лекции_1
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
From Wikipedia, the free encyclopedia
In chemical kinetics, the entropy of activation of a reaction is one of the two parameters (along with the enthalpy of activation) which are typically obtained from the temperature dependence of a reaction rate constant, when these data are analyzed using the Eyring equation of the transition state theory. The standard entropy of activation is symbolized ΔS‡ and equals the change in entropy when the reactants change from their initial state to the activated complex or transition state (Δ = change, S = entropy, ‡ = activation). It determines the preexponential factor A of the Arrhenius equation for temperature dependence of reaction rates. The relationship depends on the molecularity of the reaction: for reactions in solution and unimolecular gas reactions A = (ekBT/h) exp(ΔS‡/R), while for bimolecular gas reactions A = (e2kBT/h) (RT/p) exp(ΔS‡/R). In these equations e is the base of natural logarithms, h is the Planck constant, kB is the Boltzmann constant and T the absolute temperature. R‘ is the ideal gas constant in units of (bar·L)/(mol·K). The factor is needed because of the pressure dependence of the reaction rate. R‘ = 8.3145 × 10−2 (bar·L)/(mol·K).[1]
The value of ΔS‡ provides clues about the molecularity of the rate determining step in a reaction, i.e. the number of molecules that enter this step.[2] Positive values suggest that entropy increases upon achieving the transition state, which often indicates a dissociative mechanism in which the activated complex is loosely bound and about to dissociate. Negative values for ΔS‡ indicate that entropy decreases on forming the transition state, which often indicates an associative mechanism in which two reaction partners form a single activated complex.[3]
References[edit]
- ^ Laidler, K.J. and Meiser J.H. Physical Chemistry (Benjamin/Cummings 1982) p.381-2 ISBN 0-8053-5682-7
- ^ Laidler and Meiser p.365
- ^ James H. Espenson Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.156-160 ISBN 0-07-288362-6
Равновесная термодинамика, применимая к начальному и конечному состояниям превращения, ничего не говорит нам ни о промежуточных состояниях, через которые должна пройти система, ни, следовательно, о кинетике процесса. Чтобы преодолеть эту трудность, мы используем то, что называется «квазиравновесным подходом», т. е. принимаем, что промежуточная конфигурация между начальным и конечным состояниями также может исследоваться, как если бы она имела определенные значения термодинамических функций. Эта промежуточная конфигурация называется активированным состоянием. В этом нет ничего нового, хотя сейчас мы выражаемся несколько более формальным языком. На рис. 6.1 показаны изменения свободной энергии, сопровождающие такого рода превращение. Форма приведенной на этом рисунке кривой — простое следствие того факта, что если начальное и конечное состояния являются равновесными (стабильное и метастабильное), то их свободные энергии должны быть минимальными, а любой путь перехода из одного состояния в другое должен проходить через максимум. Высота этого максимума и есть «свободная энергия активации» — термин, который мы употребляли всегда, когда речь шла об энергетических барьерах процессов. Свободная энергия активации AGa связана с путем реакции, который не зависит от термодинамики начального и конечного состояний. Выбирается путь с наименьшим AGa. Например, различные механизмы диффузии, описанные ранее, характеризуются разными значениями AGa. Более искусственный пример — применение катализаторов с целью направить реакцию по пути с более низкой свободной энергией активации. Между движущей силой и свободной энергией активации нет прямой связи, хотя в процессах нуклеации обе они связаны со степенью переохлаждения, как было показано ранее. Более значительная степень переохлаждения понижает барьер энергии активации и увеличивает движущую силу нуклеации. Заметьте. впрочем, что большее переохлаждение снижает также и тепловую энергию атомов, что может превратить преодоление даже пониженного энергетического барьера в более трудную задачу. Вопрос о переохлаждении, оптимальном для превращения, будет обсуждаться в этой главе ниже.
Свободная энергия активации AGa связана с путем реакции, который не зависит от термодинамики начального и конечного состояний. Выбирается путь с наименьшим AGa. Например, различные механизмы диффузии, описанные ранее, характеризуются разными значениями AGa. Более искусственный пример — применение катализаторов с целью направить реакцию по пути с более низкой свободной энергией активации. Между движущей силой и свободной энергией активации нет прямой связи, хотя в процессах нуклеации обе они связаны со степенью переохлаждения, как было показано ранее. Более значительная степень переохлаждения понижает барьер энергии активации и увеличивает движущую силу нуклеации. Заметьте. впрочем, что большее переохлаждение снижает также и тепловую энергию атомов, что может превратить преодоление даже пониженного энергетического барьера в более трудную задачу. Вопрос о переохлаждении, оптимальном для превращения, будет обсуждаться в этой главе ниже.
Энтальпия и энтропия активации. Точно так же как мы определяем энтальпию H и энтропию S и далее свободную энергию Гиббса G (= H—TS) для начального и конечного состояний, эти же термодинамические функции можно определить и для активированного состояния. Энтальпия активации АНа — это, следовательно, разность между энтальпией активированного состояния и энтальпией первоначального состояния. Кривая изменения энтальпии в ходе реакции во многом такая же, что и для свободной энергии (рис. 6.1), за исключением одного важного отличия. Энтальпия конечного состояния может быть меньше или больше, чем первоначального, но свободная энергия конечного состояния всегда должна быть меньше. Если энтальпия конечного состояния меньше, то реакция эзотермична, если больше, то она эндотермична.
Аналогичным образом энтропия активации ASa — это разность энтропий активированного и первоначального состояний. В физическом смысле энтропия активированного состояния связана с множеством конфигураций активированного состояния, и здесь применимы все энтропийные члены, описанные ранее. Ее также можно представить как множество путей перехода из первоначального в конечное состояние. На практике всегда имеется более чем один набор конфигурационных изменений, способных вызвать данное превращение.
Таким образом, свободную энергию активации можно записать как
AGа = AHа — TASа.
Следовательно, активационный барьер свободной энергии при увеличении энтропии активации будет понижаться, другими словами, чем больше число возможных путей реакции, тем быстрее процесс. Примером могут служить превращения порядок — беспорядок. В процессе упорядочения существует относительно мало путей превращения разупорядоченного состояния в упорядоченное по сравнению с громадным числом возможностей для обратного процесса. В этом состоит одна из причин того, что разупорядочение является более быстрым процессом, чем упорядочение. Сходным образом испарение обычно происходит быстрее, чем конденсация, а кристаллизация — медленнее, чем плавление.
Скорость термически активированного процесса. Уравнение (5.10), описывающее температурную зависимость коэффициента диффузии, является общим для термически активированного процесса в том смысле, что оно выводится путем рассмотрения вероятности достижения отдельным атомом достаточной энергии, чтобы преодолеть активационный барьер свободной энергии. Следовательно, мы можем записать, что для любого термически активированного процесса Независимая от температуры константа const*еSa/R называется частотным фактором и обозначается буквой А. Она зависит от частоты, с которой реагирующий атом входит в активированное состояние, и, следовательно, в широком смысле от механизма процесса:
Независимая от температуры константа const*еSa/R называется частотным фактором и обозначается буквой А. Она зависит от частоты, с которой реагирующий атом входит в активированное состояние, и, следовательно, в широком смысле от механизма процесса: Это соотношение часто называют уравнением Аррениуса.
Это соотношение часто называют уравнением Аррениуса.
Если мы полагаем, что скорость процессов упорядочения или распада при охлаждении природных минералов управляется экспоненциальной функцией такого вида, то не приходится удивляться, что так часто встречаются метастабильные состояния. Например, активационная энергия упорядочения Si и Al в альбите составляет около 60 ккал/моль. Принимая R равной 2 кал/(моль*°С), находим, что при 1000 К Таким образом, при 1000 К скорость процесса в 10в31 раз быстрее, чем при комнатной температуре! При меньших энергиях активации изменение скорости с температурой не столь заметно. Это лучше всего видно на диаграмме зависимости скорости реакции от температуры:
Таким образом, при 1000 К скорость процесса в 10в31 раз быстрее, чем при комнатной температуре! При меньших энергиях активации изменение скорости с температурой не столь заметно. Это лучше всего видно на диаграмме зависимости скорости реакции от температуры: Таким образом, зависимость логарифма скорости реакции от величины, обратной температуре (1/Т), будет линейной с градиентом, равным — На/2,303R, а точка пересечения этой линии с осью скоростей будет определяться механизмом реакции. Такая диаграмма известна как диаграмма Аррениуса (рис. 6.2). Если экспериментально измеренные скорости реакции нанести на эту диаграмму, то линейный характер зависимости укажет на термически активированный механизм; кроме того, можно определить величины Hа и А.
Таким образом, зависимость логарифма скорости реакции от величины, обратной температуре (1/Т), будет линейной с градиентом, равным — На/2,303R, а точка пересечения этой линии с осью скоростей будет определяться механизмом реакции. Такая диаграмма известна как диаграмма Аррениуса (рис. 6.2). Если экспериментально измеренные скорости реакции нанести на эту диаграмму, то линейный характер зависимости укажет на термически активированный механизм; кроме того, можно определить величины Hа и А.
 На рис. 6.3,а, б показано влияние высоких и низких значений энергии активации на наклон линии на диаграмме Аррениуса. На рис. 6.3, в диаграмма Аррениуса для двух реакций с одной и той же энергией активации, но с заметно различными скоростями иллюстрирует влияние частотного фактора. Иначе говоря, реакция с самой высокой энтропией активации будет иметь самую низкую свободную энергию активации и поэтому будет протекать наиболее быстро.
На рис. 6.3,а, б показано влияние высоких и низких значений энергии активации на наклон линии на диаграмме Аррениуса. На рис. 6.3, в диаграмма Аррениуса для двух реакций с одной и той же энергией активации, но с заметно различными скоростями иллюстрирует влияние частотного фактора. Иначе говоря, реакция с самой высокой энтропией активации будет иметь самую низкую свободную энергию активации и поэтому будет протекать наиболее быстро.
В применении к скоростям диффузии экспериментальные диаграммы Аррениуса могут быть использованы для идентификации температурной зависимости различных механизмов диффузии. Например, можно ожидать, что при высоких температурах доминирующим механизмом будет объемная диффузия, тогда как при более низких температурах будет преобладать механизм с более низкой активационной энергией (например, диффузия по границам зерен). Схематическая диаграмма такого рода показана на рис. 6.4.
Энтропия — активация
Cтраница 3
Энтропия активации отрицательна и, как правило, велика.
[32]
Энтропия активации этой обменной реакции вдвое превышает энтропию активации любой другой реакции ( — 52 кал / ( град — моль), что указывает на образование более сложных переходных соединений.
[33]
Энтропия активации не зависит от предыстории пленок.
[34]
Энтропия активации, вычисленная на основании известной скорости этой реакции в нитробензоле при 60, равна — 36 1, в то время как разность энтропии образующихся и исходных веществ равна — 37 1л: а / градус. Такое совпадение означает, что активированный комплекс данной реакции по своей структуре должен быть подобен соли четырехзамещенного аммония, рассматриваемой как продукт реакции, что находится в согласии с уже сделанным предположением.
[35]
Энтропии активации этих реакций близки, однако скорости их сильно различаются. Измерения показывают, что их энергии активации различны. Но отношение скорости реакции с водородом к скорости реакции с бромом ( при одинаковых температурах и концентрациях) примерно в девять раз больше, чем получится из расчета, если мы учтем разницу в энергиях активации.
[36]
Энтропия активации или, иначе говоря, ориентационные требования для прямой и обратной реакций редко совпадают. Значит, в больших концентрациях накапливаются при равновесии те вещества, для взаимодействия которых необходимо выполнение особенно жестких требований ориентации. Катализатор может изменить энтропию активации, однако и в этом случае он не влияет на положение равновесия. Он влияет только на скорости.
[37]
Энтропия активации близка к нулю.
[38]
Энтропия активации отрицательна и, как правило, велика.
[40]
Энтропия активации дает некоторое представление о механизме элементарных реакций, так как она связана с изменением упорядоченности системы при образовании активированного комплекса. Для бимолекулярных реакций эта упорядоченность возрастает, а энтропия активации имеет отрицательное значение. Наоборот, для мономолекулярных реакций переходное состояние из-за удлинения рвущихся связей становится менее упорядоченным, и энтропия активации приобретает положительное значение.
[42]
Энтропия активации может быть меньше нуля, если активированный комплекс имеет более упорядоченную структуру, чем исходные вещества.
[43]
Энтропия активации ( отнесенная к одному пути реакции) может быть получена из экспериментального предэкспонента Ат для высоких давлений с использованием уравнения (6.7), причем вычитанием из него величины ln ( Qj 7Qi) можно при необходимости определить колебательный вклад AS в энтропию активации. Поскольку частоты колебаний молекул в общем известны, колебательная энтропия исходной молекулы SV ( A) при заданной температуре может быть рассчитана с использованием уравнений (6.9) — (6.11) для модели гармонического осциллятора. Затем следует построить модель активированного комплекса с колебательной энтропией 5j ( A) Sj ( A) ASj. На конечном этапе производится точная подгонка по одной или нескольким частотам, что делается либо вновь с помощью табл. II.
[44]
Энтропия активации, в свою очередь, тесно связана со строением исходных молекул и активного комплекса. Например, разрушение сложных молекул и образование более простого по строению активного комплекса ведет к росту беспорядка в системе и сопровождается повышением энтропии.
[45]
Страницы:
1
2
3
4
Лекция 28. Теория активированного комплекса.
Статистическая и термодинамическая формулировка.
Оглавление
Теория
абсолютных скоростей реакции
Различные
формы основного уравнения ТАК
Свободная
энергия, энтальпия и энтропия активации
Приложение
теории активированного комплекса и теории соударений к тримолекулярным реакциям
Теория абсолютных скоростей
реакции
А
формальной (феноменологической) кинетике сведения о скоростях и энергиях
активации получают непосредственно из эксперимента. В этом случае расчетные
методы сильно ограниченны.
Принципиальный
интерес представляют вариантность расчета абсолютных скоростей реакций исходя
из свойств молекул. Этот раздел химической кинетики называется теорией
абсолютных скоростей, которая включает в себя 2 задачи:
1.Нахождения
уравнения потенциальной поверхности;
2.Нахождение
связи скорости реакции с формой потенциальной поверхности.
Для
решения второй задачи Эйронгом и Поляни
был предложен метод активированного комплекса
который позволяет на основе некоторых экспериментальных параметров,
характеризующих потенциальную поверхность, рассчитать скорость реакции.
Пусть
δ – область состояний вблизи вершины потенциального барьера. Большинство
экспериментальных реакций преодолевают эту область протяженностью δ за
конечный интервал времени τ.
Допустим,
что в некоторый момент времени в системе С# активированных комплексов в единице объема
количество продуктов реакции образовавшихся в единице объема за единицу времени
совпадает со скоростью реакции:
![]()
Следовательно
задача описания кинетики сводится к нахождению С#,
τ. Время прохождения пути δ можно найти через среднюю скорость ![]() :
:
![]()
Рассмотрим
движение системы через переходное состояние как поступательное движение вдоль
координаты x.
Из
кинетической теории газов известно, что число частиц dN , движущихся со скоростью ![]() определяется по
определяется по
формуле:
![]()
![]()
Исходя из этого можно определить:


Следовательно:

При
определении С# исходят из допущения: переход активированного
комплекса в продукты не нарушает распределение Максвелла-Больцмана, т.е.
активированный комплекс расходуется быстрее, чем протекает химическое
превращение. Это допущение позволяет выразить концентрацию активированного
комплекса через концентрации реагентов, первые энергии и статические суммы
активированного комплекса и реагирующих частиц.
Активированный
комплекс можно рассматривать как квазичастицу, образованную из частиц
реагентов.
Таким
образом, исходя из статистической функции можно записать:
![]()
где СА,
СВ – концентрации А, В и т.д.
Еa – разность нулевых энергий активированного комплекса
и исходных частиц
QA, QB –
статистические суммы исходных частиц.
![]() —
—
статистическая сумма активированного комлекса.
В отличие
от других частиц активированный комплекс отличается тем, что перемещению вдоль
оси x соответствует максимум энергии:
![]()
где Q# —
статистическая сумма для совокупностей всех остальных степеней свободы.
Статистическая
сумма, характеризующая движение вдоль отрезка δ можно записать:
![]()
С учётом
этого
![]()
Отсюда

Основная
формула активированного комплекса таким образом:
![]()
При выводе
этого уравнения движения конфигуративной точки по поверхности
потенциальной энергии были положены в основу 4 постулата:
1.
Молекулярная
система движется по ППЭ в направлении от реагентов к продуктам, через седловую точку,
2.
Движение
молекулярной системы по ППЭ описывается в терминах классической мехнаики,
3.
Молекулярная
система движется по ППЭ адиабатически,
4.
Концентрация
активированного комплекса может быть выражена через термодинамическую
константу равновесия.
Обратим
внимание на третий постулат, который требует плавного изменения всех параметров
системы, т.е. плавного перемещения конфигуративной
точки вдоль ППЭ. Однако возможны и неадиабатические
скачкообразные переходы. Например в реакции:
![]()
Атом О имеет или S=1 (неспаренные электроны параллельны). При образовании СО2 состояние электрона меняется поэтому сам
процесс скачкообразный.
В неадибатических процессах и рекциях
с образованием одноя частицыпри
малых давлениях учитывается вероятность достижения переходного состояния. С
этой целью вводится коэффициент прохождения или трансмиссионный
коэффициент χ и основная формула метода активированного комплекса
записывается:
![]()
Основным
ограничением этого метода является отсутствие методов изучения активированного
комплекса. Однако теория переходного состояния или теория активированного
комплекса была огромным прорывом. Во-первых, был выработан общий
подход к количественному описанию динамики элементарного акта. Во-вторых, ТАК указан
преимущественный путь к расчету абсолютных величин констант скоростей
только на основе данных о строении реагирующих веществ.
Эта теория
позволила дать полуколичественную, или по крайней мере
качественную интерпретацию экспериментальных фактов.
Различные
формы основного уравнения ТАК
Помимо
основного выражения
![]()
можно
записать
![]()
С учетом
уравнения изотермы:
![]()
Величину ![]() можно разложить
можно разложить
на энтальпийную и энтропийную составляющую:
![]()
Отличительной
особенностью ТАК является то, что она справедлива в равной степени для
элементарных реакций любой молекулярности. Однако, возникает вопрос о размерности констант скорости. На
первый взгляд она имеет размерность с-1 (сомножитель kT/h), поскольку
экспоненты безразмерны. Однако нельзя забывать, что в показатель экспоненты входят стандартные величины
термодинамических функций в которых осуществлен выбор стандартного состояния С1=1 М.
Свободная
энергия, энтальпия и энтропия активации
Термодинамические
функции активации характеризуют процесс перестройки атомно-молекулярной
структуры при химической реакции. Очевидно, что при ![]() атомы в
атомы в
активированном комплексе менее связаны, чем в исходной молекуле.
Какие значения может принимать ![]() Очевидно она
Очевидно она
может быть рассчитана по уравнению:
![]()
Так для
реакции нейтрализации при 298,45 К, для которой k298=1,4·1011
м-1·с-1
![]()
Следует иметь ввиду, что это одна из наиболее быстрых реакций. Для быстрых реакций рекомбинации свободных радикалов ![]() . Положительная величина говорит о том, что
. Положительная величина говорит о том, что ![]() .
.
Уравнение
ТАК вносит коррективы в классическую формулировку Аррейниуса:
скорость реакции определяется не энергией активации, а свободной энергией
активации. Энтальпия активации играет важную роль лишь при![]() . Поэтому возникает вопрос
. Поэтому возникает вопрос
об оценке этой величины. Непосредственное определение ![]() затруднено
затруднено
из-за температурной зависимости.
Допустим,
что константа скорости и теоретическая константа из
теории ТАК идентичны. В этом случае аррейнуская
энергия активации:

где ![]() — теоретическая
— теоретическая
константа.
Прологарифмируем
выражение:
![]()
![]()
Найдем
энергию активации из этого выражения:

![]()
Связь ![]() выражается
выражается
![]()
Но для мономолекулярных
реакций и реакций в конденсированной фазе: ![]() , поэтому
, поэтому
![]()
Для реакции
в газовой фазе ![]() , где
, где ![]() — изменение
— изменение
числа молей при образовании переходного комплекса из исходных веществ:
![]()
![]()
Как
известно из термодинамики ![]() не зависят от
не зависят от
выбора стандартного состояния, однако этого нельзя
сказать об энтропии активации. Для неё справедливо соотношение
![]()
где x – молекулярность в стадии образования переходного комплекса.
Приложение
теории активированного комплекса и теории соударений к тримолекулярным реакциям
Рассмотрение
тримолекулярных реакций можно провести как в рамках формальной кинетики, так и
в рамках обеих теорий химической кинетики ТАС и ТАК.
В 1914 г. Траутц открыл простую тримолекулярную реакцию :
![]()
Позднее
были обнаружены и исследованы ещё 2 аналогичные реакции:
![]()
Причем для
последней реакции было обнаружено снижение константы скорости от температуры.
В 1922 г. Боденштейн предпринял попытку рассмотреть
эту реакцию в рамках JC
предложив механизм:
Быстро ![]()
Медленно ![]()
Очевидно,
что в рамках формальной кинетики
![]()
Если
стадия I экзотермична, то уменьшение скорости реакции с температурой
объяснимо. Однако, несмотря на удовлетворительное
качественное объяснение опытных данных, количественная проверка дала превышение
расчетной эффективной константы скорости в 105 раз по сравнению с
экспериментальной, т.е. согласование этих величин дало стерический
множитель в ТАС 10-5.
ТАК
позволила объяснить эту «аномалию». Так в реакции окисления оксида азота (II) можно предположть
образование комплекса с написанной структурой
![]()
В этом
случае ![]() однако в координатах
однако в координатах![]() наблюдаются заметное отклонение от линейности.
наблюдаются заметное отклонение от линейности.
«Спрямить»
эту зависимость можно, предположив вращение вокруг связи О
– О в комплексе, что понижает показатель степени у Т до -3,0, а также необходимо
учесть зависимость колебательных сумм по состоянию от температр
в виде аппроксимирующей функции f(T). В этом случае
![]()
Это даст
хорошую сходимость с экспериментом. ТАК объяснила
почему ТАС сильно занижает расчетные величины предэкспоненты.
В ТАК помимо kT/h входит ![]() .
.
Этот множитель в тримолекулярных реакциях вносит заметный
вклад. Комплекс образуется с ![]() и численном значении
и численном значении ![]() .
.