Для получения наиболее
полной информации о механизме реакции необходимо вычислить многомерную поверхность
потенциальной энергии (ППЭ), т.е. рассчитать зависимость полной энергии от координат
атомных ядер. Наиболее интересными и важными при изучении механизма реакции
являются так называемые стационарные точки на ППЭ. Под этим термином понимают
минимумы и седловые точки на ППЭ (рис. 1.4). В стационарных точках производные
полной энергии Е по всем независимым координатам хi
равны нулю:
gi= ∂E/∂xi=0 (для любого i).
В точке минимума полной
энергии матрица вторых производных fij= ∂2E/∂xi∂xjимеет только
положительные собственные значения, а в седловой точке — одно отрицательное
собственное значение. Минимумы полной энергии соответствуют устойчивым
структурам и интермедиатам, а седловые точки — переходным состояниям.
Основная трудность, с
которой мы сталкиваемся при расчете ППЭ для органических реакций с участием
многоатомных молекул, заключается в необходимости вычисления полной энергии
реагентов для очень большого числа точек.
|
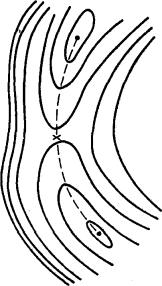
Рис. 1.4. Стационарные точки на ППЭ а — минимум локальный б — седловая точка
|
Рис. 1.5. Простейшая Темные кружки – и продукты реакции; крестик — переходное
|


Если в реагентах содержится лишь 10 атомов
(обычно их значительно больше), то их положение в пространстве для нелинейной
системы описывается 24 независимыми координатами. Для надежного построения
полной ППЭ для каждой независимой координаты необходимо взять по крайней мере
около 10 точек, таким образом полную энергию необходимо вычислить около 1024
раз. Это невероятно большая величина, и такое количество вычислений проделать
практически невозможно. Поэтому при построении ППЭ обычно варьируют не все
независимые степени свободы, а лишь некоторые из них, чаще всего один или два
геометрических параметра, которые наиболее сильно меняются в ходе реакции, а по
всем остальным степеням свободы проводят оптимизацию, т.е. находят их
оптимальные значения (соответствующие минимуму полной энергии) для каждой точки
ППЭ. Для разделения всех степеней свободы на те, которые надо варьировать, и
те, которые можно оптимизировать, используют химическую интуицию. Переход от варьирования
к оптимизации для большинства степеней свободы при построении ППЭ позволяет
очень сильно сократить затраты машинного времени и делает расчет ППЭ
практически возможным для реакций с участием достаточно больших органических
соединений.
Типичный вид простейшей двумерной ППЭ показан на
рис. 1.5. Здесь минимумы соответствуют исходным реагентам и конечным продуктам
реакции, а седловая точка — переходному состоянию. Минимумы на рисунке соединены
пунктирной линией, которая проходит по дну долины на ППЭ через седловую точку.
Эта линия показывает путь реакции в двумерном (в общем случае многомерном)
пространстве или траекторию движения реагентов в ходе реакции.
На рис. 1.5. показана простейшая ППЭ. Для
большинства реакций ППЭ имеют более сложный вид. На них может быть более двух
глубоких минимумов, которые соответствуют исходным реагентам и различным
конечным продуктам реакции (если реакция может идти по нескольким
направлениям). Кроме того, может быть несколько мелких локальных минимумов,
соответствующих интермедиатам. Реакция может идти по нескольким траекториям
через разные переходные состояния и приводить к разным конечным продуктам. При
вычислении константы скорости k = koexp(-Eакт/RT)
элементарных реакций с участием органических молекул обычно пользуются теорией
переходного состояния. В рамках этой теории для вычисления энергии активации Еакт
и предэкспоненты kо необходимо знать
потенциальную энергию, геометрию, а также полный набор колебательных частот
исходных реагентов и переходного состояния.
Для бимолекулярной реакции
А + В → АВ# → С + D
ko = kБTQ#/(hQAQB),
где
kБ и h- универсальные
постоянные; Т — температура; QA, QB
иQ# — статистические суммы исходных реагентов А и В
и переходного состояния АВ#.
Каждая из этих статистических сумм является
произведением трансляционной, вращательной и колебательной статистических сумм
Q = Qтр
Qвр Qкол;
Qтр = [mkБT/(2πh2)]3/2,
где
m — масса молекулы или переходного состояния;
Qвр = π7/2(8kБТ/h2)3/2(IAIBIC)1/2
где
IА, IB и IC—
моменты инерции;
Qкол = Пi(1
— ехр(hνi/kБТ)
где
νi — колебательные частоты (произведение
берется по всем колебательным степеням свободы).
Для вычисления энергии активации Еакт и
предэкспоненты ko по теории переходного
состояния необходимо знать геометрию, потенциальную энергию и полные наборы
колебательных частот соединений А, В и переходного состояния. Геометрия нужна
для определения моментов инерции в формуле для Qвр, значения потенциальной
энергии — для расчета Еакт, колебательные частоты — для
вычисления вклада энергии нулевых колебаний в Еакт и
колебательной статсуммы Qкол.
Таблица 1.9 Вычисленные энергии активации
(кДж/моль) для реакций, приведенных на схемах I — V
[56]
|
Реакция |
МПДП |
КМПДП |
ХФ |
КХФ* |
|
1 |
400 |
331 |
344 |
327 |
|
2 |
523 |
420 |
483 |
419 |
|
3 |
627 |
589 |
638 |
559 |
|
4 |
343 |
316 |
315 |
286 |
|
5 |
396 |
271 |
212 |
202 |
|
6 |
458 |
390 |
437 |
402 |
|
7 |
405 |
299 |
455 |
361 |
|
8 |
84 |
96 |
119 |
131 |
|
9 |
538 |
502 |
550 |
— |
|
10 |
568 |
540 |
483 |
430 |
|
11 |
458 |
382 |
411 |
436 |
|
12 |
425 |
347 |
426 |
403 |
|
13 |
453 |
323 |
348 |
378 |
|
14 |
341 |
349 |
338 |
353 |
|
15 |
318 |
268 |
278 |
262 |
|
16 |
78 |
38 |
26 |
23 |
|
17 |
360 |
201 |
218 |
161 |
|
18 |
444 |
352 |
397 |
326 |
|
19 |
609 |
467 |
538 |
428 |
|
20 |
370 |
238 |
254 |
200 |
|
21 |
399 |
372 |
452 |
390 |
|
22 |
286 |
280 |
250 |
186 |
|
23 |
403 |
310 |
420 |
— |
|
24 |
437 |
275 |
319 |
259 |
*При
расчете энергии активации использована геометрия переходного состояния,
предварительно вычисленная в приближении Хартри—Фока.
Таким образом, мы видим, что для определения
константы скорости реакции нет необходимости рассчитывать всю ППЭ. Достаточно найти
на ней лишь стационарные точки. Геометрию исходных реагентов для любой реакции
можно найти с помощью минимизации полной энергии. Эту задачу сравнительно легко
решить. Гораздо сложнее найти геометрию переходного состояния. Переходные
состояния являются седловыми точками на ППЭ. Они похожи на минимумы, поскольку
в них, как и в минимумах, градиент полной энергии по координатам равен нулю, но
их нельзя найти с помощью минимизации полной энергии. В работе [55] было
предложено искать переходные состояния с помощью минимизации квадрата модуля градиента,
т.е. минимизировать сумму квадратов производных полной энергии по всем
независимым координатам:
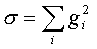
Минимизация этого выражения действительно
позволяет найти переходные состояния, однако сделать это достаточно сложно. Выражение
для σ имеет очень много минимумов, так как не только стационарные
точки дают минимумы для σ, но и любой изгиб ППЭ типа плеча приводит
к появлению минимума для этой величины. Поэтому предложенная методика [55]
пригодна лишь в непосредственной близости от седловой точки, и основная трудность
при расчете геометрии переходных состояний этим методом заключается в том, что
исходная (стартовая) геометрия, с которой начинается поиск, должна быть
достаточно близка к конечной (истинной) геометрии переходного состояния.
Определять стартовую геометрию приходится на основе интуитивных соображений,
так как из эксперимента получить хотя бы приближенную информацию о геометрии
переходных состояний невозможно. Поэтому успешный поиск переходных состояний с
помощью минимизации выражения для σ в значительной степени зависит
от опыта исследователя.
Наибольший интерес вызывает вопрос о точности, с
которой можно рассчитать геометрию переходного состояния и энергию активации
методами квантовой химии. К сожалению, мы не располагаем достаточно полным
набором данных эксперимента по энергиям активации для различных типов реакций с
участием маленьких молекул, а экспериментальные значения геометрических
параметров переходных состояний получить невозможно. Поэтому в настоящем
разделе нам пришлось ограничить рассмотрение вопроса о точности расчета
переходных состояний методами квантовой химии сопоставлением результатов,
которые были получены полуэмпирическими и неэмпирическими методами (см. [56,
57] и ссылки в них).
В табл. 1.9 и 1.10 сопоставлены данные расчета
параметров переходных состояний для реакций, изображенных на схемах I—V (см.
стр. 40), методами МПДП и КМПДП (метод МПДП с учетом электронной корреляции),
неэмпирическим методом в приближении Хартри-Фока без учета и с учетом
электронной корреляции.
В таблицах неэмпирический расчет в приближении
Хартри-Фока без учета электронной корреляции обозначен ХФ, с учетом электронной
корреляции — КХФ [56, 57].
Таблица 1.10 Статистический анализ результатов
расчета геометрии переходных состояний и энергий активации методами МПДП и
КМПДП [56, 57]
|
Вычисленная величина |
Число расчетов |
Абсолютная ошибка*1 |
|
|
МПДП |
КМПДП |
||
|
Длина валентных из них активных*3 пассивных*3 Валентный угол*2, Торсионный угол*2, Энергия активации, |
112 70 42 58 20 24 |
0,0057 0,0078 0,0018 7,9 11,6 55 90*4 |
0,0056 0,0073 0,0025 6,2 7,9 45 36*4 |
*1 По сравнению с данными
неэмпирического расчета в приближении Хартри-Фока.
*2 Для переходного состояния.
*3 Активные связи разрываются или
образуются в ходе реакции, пассивные связи в ходе реакции формально остаются
неизменными.
*4 По сравнению с данными
неэмпирических расчетов с учетом электронной корреляции.
Для реакций, изображенных на схемах I — III,
расчеты с оптимизацией геометрии в приближении Хартри-Фока проведены в базисе
6-31ГФ*, для реакций, изображенных на схемах IV и V, — в базисе 3-21ГФ или
4-31ГФ. Электронная корреляция учитывалась только при вычислении энергии активации.
Эти расчеты проведены по теории возмущений Мёллера-Плезетта с точностью до
четвертого порядка включительно или в приближении связанных электронных пар.
Схема I Схема II СхемaIII
Схема IV СхемaV

Из этих данных видно,
что геометрические параметры переходных состояний, вычисленные методами МПДП и
КМПДП, находятся в хорошем согласии с данными неэмпирических расчетов без
учета электронной корреляции. Однако для длин валентных связей, которые
формально разрываются или образуются в ходе реакции (активные связи),
расхождение результатов, полученных полуэмпирическими и неэмпирическими
методами, заметно больше, чем для валентных связей, которые в ходе реакции
остаются формально неизменными (пассивные связи). Сопоставить результаты
расчета геометрии переходных состояний методами МПДП и КМПДП с данными неэмпирических
расчетов с учетом электронной корреляции невозможно из-за почти полного
отсутствия в литературе последних. При расчете энергий активации величины,
полученные методами МПДП и КМПДП, отличаются на 15 — 20% от значений, вычисленных
неэмпирическим методом без учета электронной корреляции. При сравнении с
данными неэмпирических расчетов с учетом электронной корреляции оказывается,
что метод КМПДП дает более близкие результаты. Это, по-видимому, связано с
выбором реакций для тестирования. В отобранных реакциях учет электронной корреляции
может заметно изменить результаты расчета, так как для сравнения авторам работ
[56, 57] пришлось взять лишь те химические превращения, для которых из литературы
были известны результаты вычислений неэмпирическими методами с использованием
достаточно большого базиса и с учетом электронной корреляции. Такие расчеты,
естественно, были опубликованы лишь для тех реакций, в которых электронная
корреляция играет существенную роль. Для других типов реакций литературные данные
отсутствуют, и их получение связано с очень большим объемом вычислений.
Таблица 1.11. Вычисленные тепловой
эффект Qи энергия активации Н#
реакции (25) (кДж/моль)
|
Метод |
Q |
H# |
H#обр |
|
ХФ/3-21ГФ ХФ/6-31ГФ* МП2/6-31ГФ* МПЗ/6-31ГФ* МПЗ/6-311ГФ** МП4/6-311ГФ** С учетом нулевых |
162 144 191 178 181 193 185 |
270 213 203 207 201 202 189 |
108 69 12 31 20 9 4 |
Примечание. Использованы следующие
обозначения: ХФ — расчет в приближении Хартри-Фока; МП2, МПЗ и МП4 — расчеты с
учетом электронной корреляции во втором, во втором и третьем и во втором,
третьем и четвертом порядках теории возмущений Меллера-Плезетта. В методах
МПЗ/6-31ГФ**, МПЗ/6-311ГФ** и МП4/6-311ГФ** использована геометрия, вычисленная
предварительно методом МП2/6-31 ГФ*; Нобр — энергия активации
обратной реакции.
Некоторое представление
о влиянии базиса и электронной корреляции на результаты неэмпирических расчетов
высоты активационных барьеров можно получить из табл. 1.11 и 1.12, в которых
приведены данные для реакций
HC≡CH → H2C=C: (25),
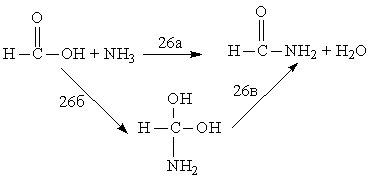
Для реакции (25)
зависимость достаточно сильная [58-60], для (26) — слабая [61]. Существенное
влияние электронной корреляции на результаты расчета высоты активационных
барьеров можно ожидать для реакций, в ходе которых образуются бирадикалы,
карбены и родственные им структуры. Кроме того, электронную корреляцию
обязательно надо учитывать при расчете ППЭ для реакций гомолитического разрыва
валентных связей.
Таблица 1.12 Энергии
активации реакций (26) (кДж/моль) [61]
|
Метод |
(26а) |
(26б) |
(26в) |
Метод |
(26а) |
(26б) |
(26в) |
|
ХФ/3-21ГФ МП2/3-21ГФ ХФ/6-31ГФ МП2/6-31ГФ ХФ/6-31ГФ* |
176 126 218 156 271 |
161 150 197 174 231 |
181 134 192 145 217 |
МП2/6-31ГФ* ХФ/6-31ГФ** МП2/6-31ГФ** МПЗ/6-31ГФ** МП4/6-31ГФ** |
180 265 176 202 197 |
167 224 163 170 174 |
165 213 162 185 176 |
Примечание. Обозначения те же, что и
в предыдущей таблице. Во всех расчетах использована геометрия молекул и переходных
состояний, вычисленная предварительно методом ХФ/3-21ГФ.
Вопрос о влиянии
электронной корреляции на геометрию переходных состояний был рассмотрен в
работе [62]. В ней методом МПДП без учета и с учетом электронной корреляции
была рассчитана геометрия переходных состояний для реакций, изображенных на
схемах I — V, и показано, что учет
электронной корреляции изменяет межатомные расстояния в среднем не более чем на
0,003 нм, при этом изменения длины активных валентных связей, т.е. тех, которые
разрываются или образуются в ходе реакции, не превышают 0,004 нм, а пассивных,
т.е. тех, которые в ходе реакции остаются формально теми же, 0,001 нм, валентные
углы меняются менее чем на 3°. Эти небольшие изменения геометрических
параметров незначительно меняют структуру переходного состояния и практически
не сказываются на результатах расчета энергии активации. На основе этих данных
в работе [62] был сделан вывод о том, что корреляционные эффекты можно не
учитывать при квантовохимических расчетах геометрии переходных состояний.
Аналогичных исследований, выполненных неэмпирическими методами, в настоящее
время нет.
From Wikipedia, the free encyclopedia
In chemistry, the transition state of a chemical reaction is a particular configuration along the reaction coordinate. It is defined as the state corresponding to the highest potential energy along this reaction coordinate.[1] It is often marked with the double dagger ‡ symbol.
As an example, the transition state shown below occurs during the SN2 reaction of bromoethane with a hydroxide anion:


The activated complex of a reaction can refer to either the transition state or to other states along the reaction coordinate between reactants and products, especially those close to the transition state.[3]
According to the transition state theory, once the reactants have passed through the transition state configuration, they always continue to form products.[3]
History of concept[edit]
The concept of a transition state has been important in many theories of the rates at which chemical reactions occur. This started with the transition state theory (also referred to as the activated complex theory), which was first developed around 1935 by Eyring, Evans and Polanyi, and introduced basic concepts in chemical kinetics that are still used today.
Explanation[edit]
A collision between reactant molecules may or may not result in a successful reaction.
The outcome depends on factors such as the relative kinetic energy, relative orientation and internal energy of the molecules.
Even if the collision partners form an activated complex they are not bound to go on and form
products, and instead the complex may fall apart back to the reactants.
Observing transition states[edit]
Because the structure of the transition state is a first-order saddle point along a potential energy surface, the population of species in a reaction that are at the transition state is negligible. Since being at a saddle point along the potential energy surface means that a force is acting along the bonds to the molecule, there will always be a lower energy structure that the transition state can decompose into. This is sometimes expressed by stating that the transition state has a fleeting existence, with species only maintaining the transition state structure for the time-scale of vibrations of chemical bonds (femtoseconds). However, cleverly manipulated spectroscopic techniques can get us as close as the timescale of the technique allows. Femtochemical IR spectroscopy was developed for that reason, and it is possible to probe molecular structure extremely close to the transition point. Often, along the reaction coordinate, reactive intermediates are present not much lower in energy from a transition state making it difficult to distinguish between the two.
Determining the geometry of a transition state[edit]
Transition state structures can be determined by searching for first-order saddle points on the potential energy surface (PES) of the chemical species of interest.[4] A first-order saddle point is a critical point of index one, that is, a position on the PES corresponding to a minimum in all directions except one. This is further described in the article geometry optimization.
The Hammond–Leffler postulate[edit]
The Hammond–Leffler postulate states that the structure of the transition state more closely resembles either the products or the starting material, depending on which is higher in enthalpy. A transition state that resembles the reactants more than the products is said to be early, while a transition state that resembles the products more than the reactants is said to be late. Thus, the Hammond–Leffler Postulate predicts a late transition state for an endothermic reaction and an early transition state for an exothermic reaction.
A dimensionless reaction coordinate that quantifies the lateness of a transition state can be used to test the validity of the Hammond–Leffler postulate for a particular reaction.[5]
The structure–correlation principle[edit]
The structure–correlation principle states that structural changes that occur along the reaction coordinate can reveal themselves in the ground state as deviations of bond distances and angles from normal values along the reaction coordinate.[6] According to this theory if one particular bond length on reaching the transition state increases then this bond is already longer in its ground state compared to a compound not sharing this transition state. One demonstration of this principle is found in the two bicyclic compounds depicted below.[7] The one on the left is a bicyclo[2.2.2]octene, which, at 200 °C, extrudes ethylene in a retro-Diels–Alder reaction.

Compared to the compound on the right (which, lacking an alkene group, is unable to give this reaction) the bridgehead carbon-carbon bond length is expected to be shorter if the theory holds, because on approaching the transition state this bond gains double bond character. For these two compounds the prediction holds up based on X-ray crystallography.
Implications for enzymatic catalysis[edit]
One way that enzymatic catalysis proceeds is by stabilizing the transition state through electrostatics. By lowering the energy of the transition state, it allows a greater population of the starting material to attain the energy needed to overcome the transition energy and proceed to product.
See also[edit]
- Transition state theory
- Transition state analogs, chemical compounds mimicking the substrate’s transition state and act as enzyme inhibitors
- Reaction intermediate
- Reactive intermediate
- Activated complex
References[edit]
- ^ Solomons, T.W. Graham & Fryhle, Craig B. (2004). Organic Chemistry (8th ed.). John Wiley & Sons, Inc. ISBN 0-471-41799-8.
- ^ The calculation used a B3LYP functional and a 6-31+G* basis set.
- ^ a b Peter Atkins and Julio de Paula, Physical Chemistry (8th ed., W.H. Freeman 2006), p.809 ISBN 0-7167-8759-8
- ^ Frank Jensen (1999). Introduction to Computational Chemistry. England: John Wiley and Sons Ltd.
- ^ Thomas A. Manz; David S. Sholl (2009). «A dimensionless reaction coordinate for quantifying the lateness of transition states». J. Comput. Chem.: NA. doi:10.1002/jcc.21440.
- ^ Buergi, Hans Beat; Dunitz, Jack D. (1983). «From crystal statics to chemical dynamics». Accounts of Chemical Research. 16 (5): 153. doi:10.1021/ar00089a002.
- ^ Goh, Yit Wooi; Danczak, Stephen M.; Lim, Tang Kuan; White, Jonathan M. (2007). «Manifestations of the Alder−Rickert Reaction in the Structures of Bicyclo[2.2.2]octadiene and Bicyclo[2.2.2]octene Derivatives». The Journal of Organic Chemistry. 72 (8): 2929–35. doi:10.1021/jo0625610. PMID 17371072.
Переходные вероятности. Матрица перехода.
Переходной
вероятностью
называют
условную вероятность того, что из
состояния
в итоге следующего испытания система
перейдет в состояние
.
Таким образом, индекс
относится к предшествующему, а
— к последующему состоянию.
Будем считать, что
число состояний конечно и равно k.
Матрицей
перехода
системы называют матрицу, которая
содержит все переходные вероятности
этой системы:

,
где
![]()
представляют вероятности перехода за
один шаг.
Отметим некоторые
особенности матрицы перехода.
-
Элементы
каждой строки матрицы представляют
собой вероятности всех возможных
переходов за один шаг из выбранного
состояния, в том числе и вероятность
отсутствия перехода (элемент строки с
равными индексами) -
Элементы
столбцов задают вероятности всех
переходов системы за один шаг в заданное
состояние -
Так
как в каждой строке матрицы помещены
вероятности событий (т.е. вероятности
перехода из состоянияв любое возможное состояние
),
которые образуют полную группу, то
сумма вероятностей этих событий равна
единице:
![]()
-
По
главной диагонали матрицы перехода
стоят вероятности

того, что система не выйдет из состояния,
а останется в нем.
Равенство Маркова
Обозначим через
![]()
вероятность того, что в результате n
шагов (испытаний) система перейдет из
состояния
в состояние
.
Например,
![]()
—
вероятность перехода за 10 шагов из
третьего состояния в шестое. Отметим,
что при n
= 1 эта вероятность сводится просто к
переходной вероятности
![]()
.
Возникает вопрос,
как, зная переходные вероятности
,
найти вероятности перехода состояния
в состояние
за n
шагов. С этой целью вводится в рассмотрение
промежуточное (между
и
) состояние r.
Другими словами, полагают, что из
первоначального состояния
за m
шагов система перейдет в промежуточное
состояние r
с вероятностью
![]()
,
после чего за оставшиеся n
– m
шагов из промежуточного состояния r
она перейдет в конечное состояние
с вероятностью
![]()
.
Используя формулу полной вероятности,
можно показать, что справедлива формула
![]()
Эту формулу называют
равенством
Маркова.
Зная все переходные
вероятности
![]()
,
т.е. зная матрицу перехода
![]()
из состояния в состояние за один шаг,
можно найти вероятности
![]()
перехода из состояние в состояние за
два шага, а значит, и саму матрицу перехода
![]()
,
далее – по известной матрице
— найти
![]()
и т.д.
Действительно,
полагая в равенстве Маркова n
= 2, m
= 1 получим
![]()
или
![]()
.
В матричном виде это можно записать как
![]()
.
Полагая n=3,
m
=2, получим
![]()
.
В общем случае справедливо соотношение
![]()
.
Пример.
Пусть матрица перехода
равна
![]()
Требуется найти
матрицу перехода
![]()
.
Умножая матрицу
саму на себя, получим
![]()
.
Для
практических применений чрезвычайно
важным является вопрос о расчете
вероятности
нахождения системы
в том или ином состоянии в конкретный
момент времени.
Решение этого вопроса требует знания
начальных условий, т.е. вероятностей
нахождения системы в определенных
состояниях в начальный момент времени.
Начальным распределением вероятностей
марковской цепи называется распределение
вероятностей состояний в начале процесса
![]()
.
Здесь через
![]()
обозначена вероятность нахождения
системы в состоянии
в начальный момент времени. В частном
случае, если начальное состояние системы
в точности известно (например
![]()
),
то начальная вероятность
![]()
,
а все остальные равны нулю.
Если
для однородной цепи Маркова заданы
начальное распределение вероятностей
и матрица перехода, то вероятности
состояний системы на n-м
шаге
![]()
вычисляются
по рекуррентной формуле
![]()
.
Для иллюстрации
приведем простой пример. Рассмотрим
процесс функционирования некоторой
системы (например, прибора). Пусть прибор
в течение одних суток может находиться
в одном из двух состояний – исправном
(![]()
)
и неисправном (![]()
).
В результате массовых наблюдений за
работой прибора составлена следующая
матрица перехода
![]()
,
где
![]()
— вероятность того, что прибор останется
в исправном состоянии;
![]()
—
вероятность перехода прибора из
исправного в неисправное состояние;
![]()
—
вероятность перехода прибора из
неисправного в исправное состояние;
![]()
—
вероятность того, что прибор останется
в состоянии «неисправен».
Пусть вектор
начальных вероятностей состояний
прибора задан соотношением
![]()
,
т.е.
![]()
(в начальный момент прибор был
неисправен). Требуется определить
вероятности состояния прибора через
трое суток.
Решение:
Используя матрицу перехода, определим
вероятности состояний после первого
шага (после первых суток):
![]()
![]()
.
Вероятности
состояний после второго шага (вторых
суток) равны
![]()
![]()
Наконец,
вероятности состояний после третьего
шага (третьих суток) равны
![]()
![]()
.
Таким образом,
вероятность того, что прибор будет
находиться в исправном состоянии равна
0,819, и того, что в неисправном –
соответственно 0,181.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
переходное состояние
Переходное состояние в химической реакции — это определенная конфигурация вдоль координаты реакции . Оно определяется как состояние, соответствующее максимальной энергии по этой координате. В этот момент, если кто-то постулирует совершенно необратимую реакцию , реагирующие частицы всегда будут стремиться к образованию продуктов [ 1 ] . Хотя это понятие можно распространить на любую физико-химическую реакцию или переход, оно используется в основном в молекулярной химии.
Концепция
Понятие переходного состояния является существенным во многих теориях, занимающихся, среди прочего, скоростями химических реакций . Это понятие было впервые разработано в теории переходного состояния (также называемой теорией активированных комплексов ) примерно в 1935 году, которая ввела основные понятия в химическую кинетику — в основном в жидких или газообразных средах — которые используются до сих пор.
Исходная концепция такова: в химической системе наличие реактивных объектов может привести к возникновению столкновений , которые могут привести (или не привести) к химической реакции.. Исход этих столкновений зависит от различных факторов, включая кинетическую энергию , ориентацию объектов друг относительно друга и их внутреннюю энергию . Даже если объекты производят то, что называется активированным комплексом , они не могут впоследствии диссоциировать с образованием продуктов реакции , при этом комплекс затем диссоциирует обратно на исходные реагенты. Эту трудность можно обойти экспериментально, направив реакцию по другому пути с помощью различных методов ( катализ и т . д .).
К этому понятию постулат Хаммондадобавляет постулат о том, что это переходное состояние близко по своей геометрической структуре либо к реагентам, либо к продуктам в соответствии с соответствующими энтальпиями присутствующих объектов.
Наблюдение за переходным состоянием
Теоретическое наблюдение
Теоретическое наблюдение переходного состояния может быть выполнено многими методами вычислительной химии в рамках нескольких теорий. Действительно, с теоретической точки зрения, переходное состояние соответствует на поверхности потенциальной энергии седловой точке первого порядка, и, таким образом, нахождение такой точки сводится к идентификации переходного состояния. Напомним, что седловая точка первого порядка соответствует минимуму во всех измерениях энергии, кроме одного.
Структуры переходного состояния могут быть найдены большинством методов квантовой химии ( DFT , MP2, …). Однако их локализация иногда проблематична, если учесть, что исходная структура должна быть относительно близкой к желаемому переходному состоянию. Действительно, в численном выражении относительно сложно выбрать «правильный» путь реакции, который может зависеть от выбранных алгоритмов минимизации энергии , выбранной физической системы и т. д. Среди методов обнаружения переходных состояний можно найти QST2 или QST3, в которых исходные структуры определяются из геометрии подложки или продуктов. Иногда проще (особенно для больших систем) провести оптимизацию полуэмпирическими квантовыми методами .как AM1 или PM3 , а затем использовать полученные геометрии в качестве исходных данных для более эффективных методов.
Экспериментальное наблюдение
Согласно правилам квантовой механики , переходное состояние нельзя «уловить» или наблюдать напрямую, потому что население этого состояния равно нулю. Однако спектроскопические методы, используемые соответствующим образом, могут привести к непосредственной близости относительно временных масштабов, в которых они применяются. Таким образом, фемтохимическая инфракрасная спектроскопия была разработана именно для этой цели, и этим методом можно идентифицировать структуры, относительно близкие к точке перехода. Однако промежуточные продукты реакцииНа пути реакции могут существовать промежуточные соединения, относительно близкие (но более стабильные) по энергии к переходному состоянию и поэтому могут затруднить идентификацию последнего.
Принцип корреляции структуры
Принцип структурной корреляции постулирует , «что изменения в структурах, происходящие вдоль координаты реакции , могут быть идентифицированы как отклонения длин связей и углов от нормальных значений основного состояния вдоль пути реакции » [ 2 ] . Согласно этому принципу, если конкретная длина связи увеличивается при достижении переходного состояния (неполное предложение — подлежит пересмотру). Демонстрация этого принципа может быть выведена из сравнения двух бициклических соединений, описанных ниже [ 3 ]. Соединение слева представляет собой бицикло[2,2,2]октен, который при 200 °C выдавливает этилен в ретрореакции Дильса-Альдера .

По сравнению с соединением справа (которое с одной алкеновой группой меньше не может вызвать эту реакцию) длина мостиковой углерод-углеродной связи теоретически должна быть меньше, потому что при приближении к переходному состоянию эта связь приобретает характер двойной связи . Для этих двух соединений предсказания проверяются по данным рентгеновской дифрактометрии и константам связи 13 C (обратная линейная зависимость от длины связи ).
Участие в ферментативном катализе
Одним из способов работы ферментативного катализа является электростатическая стабилизация переходного состояния . Уменьшение энергии этого переходного состояния позволяет большей популяции исходного материала получить энергию, необходимую для прохождения переходного состояния и, таким образом, достижения продукта.
Примечания и ссылки
- ↑ Соломонс, Т.В. Грэм и Фрайле, Крейг Б. (2004). Органическая химия (8-е изд.). John Wiley & Sons, Inc. ( ISBN 0-471-41799-8 ) .
- ↑ От статики кристаллов к химической динамике Ханс Бит Бюрги, Джек Д. Дуниц Acc. хим. Рез. ; 1983 ; 16(5); 153-161. DOI 10.1021/ar00089a002
- ↑ Проявление реакции Альдера-Риккерта в структурах бицикло[2.2.2]октадиена и производных бицикло[2.2.2]октена Йит Вуи Гох, Стивен М. Данчак, Тан Куан Лим и Джонатан М. Уайт J. Org. хим. ; 2007 ; 72(8) стр. 2929-2935; (Бумага) DOI 10.1021/jo0625610
- (ru) Эта статья частично или полностью взята из статьи Википедии на английском языке под названием « Переходное состояние » ( см. список авторов ) .
 Химический портал
Химический портал