Хроматография (ОФС.1.2.1.2.0001.15)
Государственная фармакопея 13 издание (ГФ XIII)
Хроматография — метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Взамен ст. ГФ XI, вып.1
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения. По механизму, лежащему в основе разделения, различают адсорбционную, распределительную, ионообменную и другие виды хроматографии.
В настоящее время используются следующие хроматографические методы анализа, представленные на рис.1
Рисунок 1. Методы хроматографического анализа.

Результат хроматографического разделения представляется в виде хроматограммы.
ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Хроматограмма представляет собой графическое или иное представление сигнала детектора, концентрации веществ в элюате или другой количественной величины, используемой для измерения концентрации веществ в элюате, от времени или объема подвижной фазы. В планарной (плоскостной) хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или ТСХ-пластинке (тонкослойная хроматография) последовательность зон адсорбции веществ исходной (анализируемой) смеси.
Схематически хроматограммы представляют собой последовательность гауссовых пиков на базовой линии (рис. 2).
Базовая линия – сигнал от подвижной фазы.
Пик – часть хроматограммы, регистрирующая отклик детектора. Пик отображает постепенное нарастание концентрации вещества и последующее ее уменьшение. В случае линейной изотермы сорбции кривая, описывающая пик, приближается к кривой гауссова распределения.
Основание пика — продолжение базовой линии, соединяющее начало и конец пика.
Площадь пика (S) – площадь хроматограммы, заключенная между кривой, описывающей пик, и его основанием.
Высота пика (H) – расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора.

Рисунок 2. Хроматограмма и основные хроматографические параметры: 1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося вещества; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)
Интерпретация хроматографических данных
Время удерживания (tR или t) – время, необходимое для элюирования вещества. Соответствует времени появления максимума пика на хроматограмме.
Объем удерживания (VR) – объем подвижной фазы, необходимый для элюирования вещества. Может быть вычислен по времени удерживания и скорости потока (F).
VR = F · tR
Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока.
Время удерживания несорбирующегося вещества (t0 или tм) – время, необходимое для элюирования неудерживаемого на сорбенте вещества. В эксклюзионной хроматографии t0 соответствует времени удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Объем удерживания несорбирующегося вещества (V0) – объем подвижной фазы, необходимый для элюирования неудерживаемого вещества. Может быть вычислен по времени удерживания неудерживаемого вещества и скорости потока (F):
V0 = F · t0
В эксклюзионной хроматографии V0 соответствует объему удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Общее время удерживания подвижной фазы (tt) – в эксклюзионной хроматографии время удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Общий объем удерживания подвижной фазы (Vt) – в эксклюзионной хроматографии объем удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Константа (коэффициент) распределения (K0) – в эксклюзионной хроматографии характеристика элюирования вещества из определенной колонки, которую рассчитывают с помощью выражения:

Приведенное (исправленное) время удерживания вещества (t´R) ‒ время удерживания вещества за вычетом времени удерживания несорбируемого вещества. Может быть рассчитано по формуле:
t´R = tR — t0
Исправленное время удерживания не зависит от объема трубопроводов хроматографической системы, установленных между инжектором и колонкой.
Относительное время удерживания (r) – относительное приведенное (исправленное) время удерживания вещества 2 по веществу 1:

Нескорректированное относительное время удерживания (RRT) – относительное время удерживания вещества 2 по веществу 1:

Если не указано иное, значения относительного времени удерживания, приведенные в фармакопейных статьях, соответствуют нескорректированному относительному времени удерживания
Коэффициент емкости (k´) – коэффициент емкости колонки по веществу с временем удерживания tR, показывающий, во сколько раз исправленное время удерживания вещества больше, чем время удерживания несорбируемого вещества.

Эффективность хроматографической системы ‒ параметр, характеризующий степень размывания хроматографического пика. Эффективность выражается числом теоретических тарелок (N):

W – ширина пика у основания;
W0,5 – ширина пика на половине высоты.
При расчете числа теоретических тарелок значения времени удерживания и ширины пика должны быть приведены к одинаковой размерности.
Число теоретических тарелок зависит от природы определяемого вещества, его концентрации или объема, вводимого в систему, от колонки, температуры колонки и состава подвижной фазы.
Если не указано иное, то эффективность хроматографической системы, требования к которой приведено в фармакопейной статье, рассчитывается по формуле, использующей ширину пика на половине его высоты.
Фактор асимметрии (фактор симметрии) пика (As) рассчитывают по формуле:
 где
где
W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты (рис. 3).
Если фактор асимметрии равен 1, то пик симметричен. Если фактор асимметрии больше 1, то это означает, что растянут задний фронт пика. Если фактор асимметрии меньше 1, то пик растянут спереди.

Рисунок 3. Схема расчета фактора асимметрии пика
Разрешение (Rs).
Разрешение между пиками двух веществ смеси элюирующимися друг за другом рассчитывают по формулам:

при этом tR2 ≥ tR1. При расчете разрешения величины времени удерживания и ширины пиков должны быть приведены к одинаковой размерности.
Если не указано иное, то разрешение, требования к которому приведены в фармакопейной статье, рассчитывается по формуле, использующей ширину пиков на половине высоты.
В случае, если пики несимметричны и если интенсивность пиков значительно различается, параметр Rs не всегда корректно описывает разделение хроматографических пиков. Таким образом, даже при значениях Rs ≥ 1,5 может наблюдаться неполное разделение пиков. В этих случаях при оценке разделяющей способности можно заменить параметр Rs на параметр «отношение максимум/минимум».
Отношение максимум/минимум (p/v), называемое также отношением «peak-to-valley», «пик – долина». Этот параметр позволяет оценить разделительную способность хроматографической системы. Значение p/v рассчитывается по формуле:
p/v = Hp / Hv
Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии (рис. 4).

Рисунок 4. Хроматограмма не полностью разделяемых веществ
Данное соотношение применяется для оценки разделительной способности хроматографической системы, если вещества смеси разделяются не полностью. Рассчитанное отношение максимум/минимум в значительной степени зависит от выбранного варианта интегрирования хроматограммы. Результаты измерения соотношения p/v будут некорректны в случае разметки меньшего пика методом экстраполяции смещенной базовой линии (методом тангенциальной касательной).
Отношение сигнал/шум (S/N).
Краткосрочный шум сигнала детектора (шум базовой линии) влияет на прецизионность количественного определения. Отношение сигнал/шум рассчитывают по формуле:

Н – высота пика, соответствующего рассматриваемому веществу на хроматограмме указываемого стандартного раствора, измеренная от максимума пика до экстраполированной базовой линии. Экстраполяция базовой линии проводится для сигнала на участке базовой линии во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте;
h – размах фонового шума, измеряемый либо на хроматограмме контрольного (холостого) раствора (или раствора плацебо), либо на хроматограмме того же раствора стандартного образца.
Измерение размаха фонового шума проводится во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте, расположенном, если это возможно, равномерно по обе стороны от места возможного обнаружения пика. В случае использования для измерения шума хроматограммы контрольного (холостого) раствора (или раствора плацебо) измерение размаха фонового шума проводится во временном интервале, включающем в себя время удерживания рассматриваемого вещества, при этом продолжительность временного интервала, в котором проводится измерение шума, должна не менее чем в 5 раз превышать ширину на половине высоты для пика на хроматограмме указываемого раствора стандартного образца (рис. 5).

Рисунок 5. Вычисление отношения сигнал/шум.
Объем задержки (D) (объем задержки градиента) представляет собой объем системы между точкой, в которой происходит смешение элюентов, и началом колонки.
Объем задержки градиента влияет на времена удерживания веществ и общий профиль наблюдаемой хроматографической картины, получаемой при использовании градиентного элюирования. Объем задержки хроматографической системы определяется следующим способом.
Колонка. Заменяют хроматографическую колонку капилляром (например, 1 м × 0,12 мм).
Подвижная фаза.
Подвижная фаза A — вода.
Подвижная фаза B — 0,1 % (об/об) раствор ацетона.
Скорость потока. Необходимая для создания давления, достаточного для стабильной работы насоса (например, 2 мл/мин).

Детектор. Спектрофотометрический при длине волны 265 нм.
Определяют время (t0,5) в минутах, когда оптическая плотность увеличилась на 50 %. Вычисляют объем задержки:


Рисунок 6. Определение объема задержки градиента
Прецизионность системы в условиях повторяемости
Прецизионность системы в условиях повторяемости выражается в виде рассчитанного относительного стандартного отклонения в процентах [RSD (%)] по результатам последовательных измерений не менее чем 3 вколов или нанесений раствора сравнения и рассчитывается по формуле:

В планарной хроматографии аналогом времени удерживания является фактор удерживания (Rf):
Rf = a / b
где a – расстояние от точки нанесения пробы до центра пятна, характеризующего зону адсорбции;
b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют условия хроматографирования. Оценкой хроматографической подвижности, менее чувствительной к влиянию отклонений в условиях проведения эксперимента, является величина Rst, представляющая собой отношение величины Rf одного вещества к величине Rf другого, принятого за стандарт:


Рисунок 7. Схема определения значений Rf и Rst.
Данные планарной хроматографии могут быть представлены в виде денситограмм.
РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют 4 основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
1. Метод нормирования (метод внутренней нормализации). Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.

Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле:
 где
где
Сi и С0 – концентрация i-го вещества и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-го вещества и стандартного вещества соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8 – 1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:

При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8 – 1,2.
2. Метод внешнего стандарта. Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах раствора стандартного образца.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:

S и S0 – средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов соответственно;
С и С0 – концентрации определяемого и стандартного растворов соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве раствора стандартного образца для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в растворе стандартного образца по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8 — 1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой, в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
3. Метод внутреннего стандарта. Концентрацию определяемого вещества определяют путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме раствора стандартного образца. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле:

4. Метод стандартных добавок. В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условиях с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:

Сстд — концентрация стандартной добавки;
Sx — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
Если не указано иное, в испытаниях на содержание примесей в качестве порога игнорирования (неучитываемый предел, уровень содержания вещества, при котором и при меньшем содержании пик не принимается во внимание) принимают величину 0,05 % от содержания основного вещества.
В случаях, когда при испытании на примеси требуется определение суммарного содержания примесей или количественного определения примеси, при интерпретации хроматограммы важно выбрать подходящие условия интегрирования и значения порога интегрирования [интенсивность сигнала пика соединения (высоты или площади)], при котором (и при меньшем) пик не учитывается системой сбора данных и не участвует в количественных расчетах.
С целью разметки на хроматограмме всех пиков, подлежащих учету, заданный порог интегрирования системы сбора данных не должен превышать половину порога игнорирования.
Интегрирование площади пика любой примеси, пик которой не полностью разделяется с основным пиком, предпочтительно проводить экстраполяцией смещенной базовой линии (методом тангенциальной касательной). Использование других способов интегрирования должно быть специально оговорено в фармакопейной статье.
ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
Испытания пригодности системы являются неотъемлемой частью методики и используются для того, чтобы убедиться в надлежащем функционировании хроматографической системы и обеспечить выполнение предъявляемых к ней требований.
При проведении испытаний используемое оборудование должно быть квалифицировано и способно к функционированию надлежащим образом.
Для оценки пригодности системы указывают:
- требования к параметрам, характеризующим форму пика [эффективность хроматографической системы N и фактор асимметрии (фактор симметрии) As];
- требования к разделительной способности (разрешение между пиками RS или отношение пик — долина p/v);
- требования к воспроизводимости (относительное стандартное отклонение, RSD) значений площади или высоты пиков, а также времен удерживания пиков в случае оценки подлинности соединений;
- требования к чувствительности при проведении испытания на примеси [минимально определяемая концентрация (фактический предел количественного определения) примеси]. Оценивается посредством расчета отношения сигнал/шум для раствора соответствующей концентрации.
Если в фармакопейной статье не указано иное, должны выполняться следующие требования и все дополнительные требования, приведенные в нормативном документе:
- в испытаниях на примеси или количественное содержание для пика на хроматограмме растворе стандартного образца, используемого для количественных определений, значение величины фактора асимметрии (фактора симметрии) As должно находиться в пределах от 0,8 до 1,5;
- разрешение между пиками Rs ³ 1,5 (в случае не полностью разделенных пиков вместо разрешения может быть использовано соотношение p/v. При этом требования к минимальному значению соотношения p/v устанавливаются в фармакопейной статье);
- отношение сигнал/шум для пика вещества, полученное для раствора с концентрацией, равной требуемому уровню минимально определяемой концентрации, должно быть не менее 10. Требуемый уровень минимально определяемой концентрации [фактический предел количественного определения (ПКО)] зависит от того, предполагает ли методика вычисление содержания примесей, или только полуколичественную оценку, когда результат представляется в виде «менее Х» или «не более Х», где Х – допустимое содержание примеси. Для методик, предполагающих вычисление содержания примесей, минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать значение порога игнорирования (если не указано иное – 0,05 % относительно концентрации основного вещества в испытуемом растворе). Для полуколичественных методик минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать максимально допустимое содержание примеси. При необходимости оценки содержания нескольких примесей требуемый уровень минимальной определяемой концентрация для применяемой хроматографической системы определяется примесью, нормы содержания которой наиболее строги. Если в фармакопейной статье не указано иное, то раствор вещества минимально определяемой концентрации для оценки чувствительности детектирования можно приготовить растворением стандартного образца вещества в том же растворителе, который используется для приготовления испытуемого раствора, с уровнем концентрации 0,05 % относительно концентрации основного вещества в испытуемом растворе.
Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
При количественном определении основного вещества в фармацевтических субстанциях при содержании основного вещества, близком к 100 %, максимально допустимое относительное стандартное отклонение значений интенсивности (площади или высоты) пика основного вещества для заданного количества повторных введений стандартного раствора RSDmax вычисляется по формуле:
Если в фармакопейной статье не указано иное, то значение RSD не должно превышать величин RSDmax, приведенных в таблице. Данное требование не применяется при испытаниях на примеси.

Таблица. Максимально допустимое относительное стандартное отклонение RSDmax в зависимости от верхнего предела содержания основного вещества и числа отдельных введений проб
| В, % | Число отдельных введений проб (вколов) | |||
| 3 | 4 | 5 | 6 | |
| Максимально допустимое относительное стандартное отклонение RSDmax | ||||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
Соответствие критериям пригодности системы должно поддерживаться на протяжении всего испытания. В зависимости от различных факторов, например, частоты использования методики и опыта работы с хроматографической системой, аналитик выбирает соответствующую схему проверки, подтверждающую соответствие этим требованиям.
В планарной хроматографии при проведении испытаний, регламентирующих наличие посторонних примесей/родственных соединений, должно быть предусмотрено определение предела обнаружения определяемых примесей и разделительной способности хроматографической системы.
Для подтверждения пригодности системы возможно одновременное использование двух тестов: «Подтверждение разделительной способности» и «Подтверждение чувствительности».
Тест «Подтверждение разделительной способности» проводят путём нанесения на пластинку и последующего хроматографирования специального стандартного раствора, содержащего два или более веществ с известными значениями Rf. После хроматографирования эти вещества должны разделиться, образуя зоны адсорбции, значения Rf которых равны заданным.
Тест «Подтверждение чувствительности» позволяет оценить чувствительность проявления. Он заключается в нанесении определённого количества стандартного вещества, хроматографируемого в условиях предыдущего теста и проявляемого тем же реагентом. Зона адсорбции стандартного вещества должно чётко обнаруживаться.
В каждом случае природа стандартных веществ, состав стандартного раствора, наносимые количества, приблизительные значения Rf устанавливают в фармакопейных статьях.
При необходимости для достижения требуемых критериев пригодности хроматографической системы проводится корректировка хроматографических условий.
КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
Далее приводятся пределы возможных изменений различных параметров хроматографической методики, которые могут быть внесены в нее для соответствия требованиям пригодности системы без принципиального изменения методик. Корректировка условий градиентного элюирования является более критичной, чем изократических условий, так как может вызвать сдвиг пиков на другую стадию градиента, и, таким образом, привести к некорректной идентификации пиков, наложению пиков или сдвигов, при которых элюирование интересующих соединений происходит после указанного времени регистрации хроматограммы. При внесении изменений, отличных от приведенных ниже, требуется проведение повторной валидации методики.
Проверка пригодности системы должна быть включена в методики для подтверждения получения разделения, требуемого для надлежащего проведения испытания. Тем не менее, поскольку неподвижные фазы описаны только в общем виде, и существует огромное количество доступных коммерческих фаз, отличающихся по хроматографическому поведению, некоторая корректировка условий хроматографирования может потребоваться для выполнения требований пригодности системы. В методиках с использованием обращенно-фазовой жидкостной хроматографии корректировки различных параметров не всегда приводят к удовлетворительному разделению. В этом случае может возникнуть необходимость замены одной колонки на другую, такого же типа, обеспечивающую желаемое хроматографическое поведение.
Если в фармакопейной статье не указано иное, допустимы следующие изменения параметров хроматографической системы.
Тонкослойная и бумажная хроматография
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
Пример 1: для вещества, содержание которого составляет 10 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 7 – 13 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению абсолютного содержания до 8 – 12 %. Допустимое изменение по относительному содержанию больше, чем допустимое изменение по абсолютному содержанию. Таким образом, допускается изменение содержания 7 – 13%.
Пример 2: для вещества, содержание которого составляет 5 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 3,5 – 6,5 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению его абсолютного содержания в подвижной фазе до 3 – 7 %. В этом примере допустимое изменение по абсолютному содержанию больше, чем допустимое изменение по относительному содержанию, и допустимым диапазоном содержания является 3 – 7 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Наносимый объём. При использовании пластинок с малым размером частиц сорбента (2 — 10 мкм) наносимый объём может быть изменен на 10 –20 % от заявленного объёма.
Жидкостная хроматография: изократическое элюирование
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Скорость потока подвижной фазы: ± 50 %, большая степень изменений допустима при одновременном изменении размеров колонки. При необходимости одновременного изменения размеров колонки и скорости потока сначала рассчитывается номинальная скорость потока для колонки, размеры которой отличаются от приведенной в нормативном документе, а затем допускается корректировка полученного значения скорости потока на ± 50 %.
Параметры колонки
Неподвижная фаза:
- замена типа неподвижной фазы недопустима (например, недопустима замена фазы С18 на фазу С8);
- размер частиц: максимальное уменьшение размера частиц 50 %, увеличение не допускается.
Размеры колонки
Длина колонки: ± 70 %,
внутренний диаметр колонки: ± 25 %.
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1 — внутренний диаметр колонки, указанный в нормативном документе, мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура: ± 10 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы (минимально определяемая концентрация) и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Жидкостная хроматография: градиентное элюирование
Изменение хроматографических условий для систем с градиентом требует большей осторожности, чем для изократических.
Состав подвижной фазы/профиль градиентного элюирования: незначительные изменения состава подвижной фазы и системы градиента являются приемлемыми при условии, что:
- выполнены требования пригодности системы;
- основной пик элюируется в пределах ± 15 % от обозначенного времени удерживания;
- элюирующая способность конечного состава подвижной фазы не менее предписанного состава.
Если при использовании градиентного элюирования не может быть достигнуто соответствие требованиям пригодности системы, рекомендуется оценить объем задержки градиента или сменить используемую колонку.
Объем задержки градиента. Конфигурация используемого оборудования может значительно изменить разрешение, указанные абсолютные и относительные времена удерживания. Это может произойти из-за отличия объема задержки градиента используемой системы от объема задержки градиента системы, с помощью которой проводилась валидация методики. В нормативном документе часто перед началом программы градиента включена изократическая стадия, чтобы можно было адаптировать систему к временным точкам градиента с учетом разницы объема задержки у системы, использованной для разработки методики, и у фактически использующейся системы. Решение о необходимости адаптации длительности изократической стадии для данного аналитического оборудования принимается пользователем. Если объем задержки, использованный при разработке методики, приведен в нормативном документе, то интервалы времени (t), указанные в таблице градиента, можно заменить адаптированными интервалами времени (tc), рассчитанными с помощью следующего уравнения:

D – объем задержки, мл;
D0 – объем задержки, использованный при разработке методики, мл;
F – скорость потока, мл/мин.
Изократическая стадия, введенная с этой целью, может быть исключена, если имеются данные валидации по применению методики без этой стадии.
pH среды водного компонента подвижной фазы. Изменения не допускаются.
Концентрация солей в буферном веществе подвижной фазы. Изменения не допускаются.
Скорость потока. Изменения допустимы при изменении размеров колонки.
Параметры колонки
Неподвижная фаза:
- замена типа неподвижной фазы недопустима (например, недопустима замена С18 на С8);
- размер частиц: изменения не допускаются;
Размеры колонки
- длина колонки: ± 70 %;
- внутренний диаметр колонки: ± 25 %;
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

- F1 — скорость потока, указанная в нормативном документе, мл/мин;
- F2 — скорректированная скорость потока, мл/мин;
- l1 — длина колонки, указанная в нормативном документе, мм;
- l2 — длина используемой колонки, мм;
- d1— внутренний диаметр колонки, указанный в нормативном документе мм;
- d2 — внутренний диаметр используемой колонки, мм.
Температура. ± 5 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что детектирование (предел количественного определения) и сходимость отклика (RSD площадей или высот) для пика (пиков) определяемых соединений остаются удовлетворительными.
Газовая хроматография
Параметры колонки
Неподвижная фаза:
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки);
- толщина плёнки: от минус 50 % до плюс 100 % (капиллярные колонки).
Размеры колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 50 %.
Скорость потока: ± 50 %.
Температура: ± 10 %.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Сверхкритическая флюидная хроматография
Состав подвижной фазы. Для набивных колонок количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше; для систем с капиллярной колонкой изменения не допускаются.
Длина волны детектора. Изменения не допускаются.
Параметры колонки
Неподвижная фаза:
- размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки).
Размер колонки
- Длина колонки: ± 70 %;
- Внутренний диаметр колонки: ± 25 % (набивные колонки), ± 50 % (капиллярные колонки).
Скорость потока: ± 50 %.
Температура: ± 5 %, если температура указана в нормативном документе.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
131
ными при анализе. Таким образом, необходимо научиться переводить параметр «расстояние удерживания» в параметр «время удерживания».
По указанию преподавателя представьте некоторые данные в виде параметра− «объем удерживания».
Заполните таблицу. Напишите на хроматограмме исходной и анализируемой смеси наименования компонентов. Хроматограммы приложите к отчету:
Таблица − Параметры качественного анализа
|
tR, с |
t’R, с |
l’R, мм |
V’R, мл |
lgV’R |
Порядок |
||
|
Вещество |
выхода |
||||||
|
компонентов |
|||||||
|
1. |
|||||||
|
2. |
|||||||
|
3. |
|||||||
|
Искусственная |
Х1 |
||||||
|
смесь |
Х2 |
||||||
|
Х3 |
На основании полученных данных для смесей известного состава построить график зависимости логарифмов приведенных объемов удерживания на фазе карбовакс-6000 (полярная) от логарифмов приведенных объемов удерживания. Возможно использование и других НЖФ. С помощью построенного графика провести идентификацию анализируемых неизвестных соединений.
Отразите в выводе результаты полученного исследования.
Практическая работа № 5
Количественный анализ смеси различными методами
ЦЕЛЬ РАБОТЫ: Определить количественный состав анализируемой смеси двумя методами (по указанию преподавателя).
Аппаратура, условия и объекты хроматографирования (параметры хроматографирования могут быть изменены):
Хроматограф «Цвет-530» или другой с ДИП. Насадочная колонка
|
(100х0,3) см. |
|
|
Газ-носитель |
– аргон |
|
Температура термостата колонок |
– 100° С |
|
Температура термостата испарителя |
– 130° С |

132
|
Температура термостата ДИП |
– 130° С |
||
|
Скорость газа-носителя |
– 10 мл/мин |
||
|
Шкала чувствительности детектора |
|||
|
(подбирается экспериментально) |
– 128х109 |
||
|
Объем инжектирования |
– 0,4 |
мкл |
|
|
Скорость ленты самописца |
– 0,6 |
см/мин |
|
|
Сорбент |
– инертон AW-DMXC (0,16 – 0,12 мм) |
||
|
Неподвижная жидкая фаза |
– карбовакс-6000 (10%) |
Объект анализа: Смесь, использованная в предыдущей работе или новая, выданная преподавателем.
Объект хроматографирования: 1% или 3% или 5% растворы этилового спирта, в качестве внутреннего стандарта используется 4% н— пропиловый спирт. По указанию преподавателя исследуемые растворы могут быть заменены.
Ход работы:
Записываются три хроматограммы анализируемой смеси. Вычисляют значения площадей пиков по формуле (55). Необходимые дополнительные построения выполняют тонко отточенным карандашом. При измерении ширины пиков пользуются измерительной лупой и при этом учитывают ширину линии, записываемой пером регистратора. Данные для трех хроматограмм заносят в таблицу.
Таблица 1 − Количественный анализ
|
Соединение |
h, мм |
b0,5, мм |
S, мм2 |
n |
SW |
На основании полученных данных рассчитывают воспроизводимость измерения высот и площадей пиков.
Коэффициент вариации вычисляется по формуле: n = SxWi 100%
где Sw – средняя квадратичная погрешность определения параметра пика,
Sw = W Kw
где W – разность между наибольшим и наименьшим значениями ряда измерений; Кw – статистический фактор, зависящий от числа измерений (см. табл. 2).
133
Таблица 2. Значения фактора Кw для расчета приближенного стандартного отклонения
|
Число параллель- |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
|
ных определений |
||||||||||
|
Kw |
0,89 |
0,59 |
0,49 |
0,43 |
0,40 |
0,37 |
0,35 |
0,34 |
0,33 |
Концентрацию веществ в смеси определите одним из предлагаемых ниже методов.
Анализ контрольной смеси методом внутренней нормализации
Вводится в хроматограф проба анализируемой смеси, содержащей известное число компонентов (n), измеряют площади всех пиков.
С помощью массовых коэффициентов отклика рассчитываются скорректированные площади пиков отдельных компонентов fi Si.
Массовую долю компонента (i) рассчитываем по формуле:
i= 1
В случае углеводородов и детектора ДИП для fi справедливо равенство:
|
M |
|||
|
fi = |
i |
, |
|
|
∑ C |
i |
||
|
где Mi – молекулярная масса компонента i, |
∑ Ci – число углерода в молекуле |
||
|
этого компонента. |
Пример: Дана пропан-бутановая смесь. Рассчитайте массовую долю пропана, исходя из следующих данных: в анализируемом газе были идентифицированы этан, пропан, изобутан, бутан, изопентан и пентан. Площади пиков на хроматограмме соответственно раны (в мм2): 24, 1520, 336, 1280, 125 и 18.
Решение:
|
44,10 |
1520 |
|||||||||||||
|
Wпропан = |
3 |
= 0,4631 |
||||||||||||
|
30,07 |
24 + |
44,10 |
1520 + |
58,12 |
336 + |
58,12 |
1280 + |
72,15 |
125 + |
72,15 |
18 |
|||
|
2 |
3 |
4 |
4 |
5 |
5 |
|||||||||
134
Анализ контрольной смеси методом внутреннего стандарта
В пробу вводят раствор стандарта известной концентрации, смесь вводится в хроматограф и сравниваются площади пиков вещества (i) и стандарта (s).
Аналитическое определение проводят следующим образом: Смешивают определенный объем анализируемой пробы (Vi) с определенным объемом (Vs) раствора стандарта (s) известной концентрации Cm,s.
Определенный объем смеси вводится в хроматограф, измеряются площади пиков вещества (i) и вещества стандарта (s) по хроматограммам
Si и Ss.
Концентрацию Cm,i вещества i рассчитывают по формуле:
|
Cm,i = |
Vs fi Si |
Cm,s |
|
Vi fs Ss |
Величину fi/fs определяют как отношение площадей модельной смеси с известными концентрациями Cm,i = Cm,s.
Пример: Данным методом с ДИП с использованием внутреннего стандарта в крови был определен этанол, рассчитать его массовую концентрацию.
В качестве внутреннего стандарта применялся метилэтилкетон. Величину fi/fs определяли при анализе водного раствора этанола (i) и метилэтилкетона (s) одинаковых концентраций Cm,i = Ci,s = 2 мг/мл получили значения: Si = 401 мм2 и Ss = 859 мм2. Тогда из вышеприведенного
уравнения следует, что fi/fs = 2,142 (отношение Vs Cm,s/Vi Cm,i = 1). Собственно анализом крови были получены следующие данные: объем крови
Vi = 1 мл, объем водного раствора стандарта с концентрацией
Cm,s = 1,014 мг/мл, Vs = 1 мл, Si = 425 мм2 и Ss = 615мм2.
Решение:
|
Cm (этанол) = |
1 2,142 425 |
1,014 |
= 1,501 мг/мл |
|
1 615 |
Возможно определение концентрации (С) данным методом используя калибровочные коэффициенты (К), данный метод описан в главе
V.
Можно построить калибровочный график зависимости: отношения площади исследуемого вещества к площади пика стандарта от концентрации определяемого вещества.
Полученные данные оформляются в таблице 3.
|
135 |
|||||||||||||||
|
Таблица 3 − |
Результаты эксперимента |
||||||||||||||
|
Исследуемое вещество |
Стандарт |
Sв− ва |
|||||||||||||
|
С0,5 |
К |
||||||||||||||
|
m, |
h, |
b0,5, |
S, |
m, |
h, |
b0,5, |
S, |
Sст |
|||||||
|
% |
мм |
мм |
мм2 |
% |
мм |
мм |
мм2 |
||||||||
|
1 |
|||||||||||||||
|
2 |
|||||||||||||||
|
3 |
|||||||||||||||
|
Х |
|||||||||||||||
|
К = mi Sст / mст Si |
mn = Kст Sст / Cст |
||||||||||||||
|
Cx = Kст Sст / m |
S = C0,5 h b0,5 |
Анализ контрольной смеси методом стандартной добавки
К исследуемой пробе добавляют определенное количество анализируемого вещества (i). Проводится два ввода пробы: в первый раз вводится определенное количество исходной пробы, а во второй раз определенное количество смеси, в которую введена стандартная добавка. Последовательность операций:
1. В хроматограф вводится определенный объем (Vi) анализируемой пробы, измеряется площадь Si площадь пика вещества (i) на хроматограмме.
2.Определенный объем анализируемой пробы (V0i) смешаем с определенным объемом (V0s) раствора определяемого вещества (i = s) известной концентрации (Cs).
3.В хроматограф вводится определенный объем (V’i) смеси, по-
|
лученной по п. 2, измеряется площадь (S’i) |
площадь пика вещества (i) |
|||||||||
|
на хроматограмме. |
||||||||||
|
4. Концентрацию (Cm,i) вещества (i) в исходной пробе рассчиты- |
||||||||||
|
вают по формуле: |
− 1 |
|||||||||
|
Vs0 |
Cm,s |
S’ |
V |
V0 |
||||||
|
Cm,i = |
i |
i |
1+ |
s |
− 1 |
|||||
|
V0 |
Si V’i |
V0 |
||||||||
|
i |
i |
Пример: Методом стандартной добавки идентифицирован бензол в нитроразбавителе. Рассчитать его концентрацию Cm,i на основании следующих данных: объем введенной пробы — Vi = 1 мкл, Si = 865 мм2, V0i = 10 мл, V0s = 5 мкл, стандарт добавлен в виде чистого (неразбавленного) бензола, поэтому Cm,s тождественна его плотности, т.е.
Cm,s = ρ = 0,879 г/мл. После ввода новой пробы получаем: V’i = 1 мкл, S’i = 1040 мм2.

136
Решение: В нашем случае V0s/V0i << 1, поэтому этим соотношением можно пренебречь. Тогда получим:
|
5 10− 3 |
0,879 |
1040 |
1 |
− 1 |
− 3 |
||||||||
|
Cm (бензол) = |
− |
1 |
= 2,17 10 |
г/мл |
|||||||||
|
10 |
867 |
1 |
|||||||||||
Анализ контрольной смеси методом абсолютной калибровки
Существует два варианта: прямое сравнение и метод калибровочной кривой.
В случае прямого сравнения поступают следующим образом:
1.Вводится в хроматограф определенный объем (Vi) анализируемой пробы, измеряется площадь пика вещества (i) т.е. Si, концентрацию которого необходимо определить.
2.В абсолютно тех же условиях хроматографирования вводим
определенный объем (Vs) калибровочного раствора с известной концентрацией стандарта (Cm,s), измеряют Ss, т.е. площадь пика вещества (s).
3.Для расчета концентрации вещества (i) в анализируемой пробе используют уравнение:
f S V
Cm,i = fs Ss Vi Cm,s
Вслучае, если калибровать стандартным раствором анализируемо-
го вещества (i). Тогда i = s, исключается необходимость определения соотношения fi / fs и уравнение упрощается:si i
Методом калибровочной кривой пользуются очень часто при серийных анализах.
Cm,i = Si Vs Cm,s
Ss Vi
С этой целью готовят растворы анализируемого вещества различной концентрации с учетом того, чтобы она не выходила за линейный динамический диапазон детектора.
Вводят в хроматограф определенное количество пробы, измеряют площади полученных пиков и строят калибровочную прямую зависимости концентрации вещества (Ci) от площади (Si).
После чего от преподавателя получают вещество неизвестной концентрации, хроматографируют его в тех же условиях и определяют его концентрацию.
Отразите результаты полученного исследования в выводах.

137
Практическая работа № 6
Газохроматографический анализ моносахаридов [10]
ЦЕЛЬ РАБОТЫ: Определить качественный и количественный состав гидролизатов или изолированных моносахаридов.
В гидролизатах происходит мутаротация моносахаридов, т.е. образование равновесной смеси таутомерных форм: α -, β -пиранозных, α -, β — фуранозных и открытой (альдегидной). Для глюкозы это иллюстрирует следующая схема:
|
CH2OH |
CH2OH |
||||
|
OH |
O |
O OH |
|||
|
H |
O |
OH |
|||
|
HO |
OH |
HO |
|||
|
C |
|||||
|
OH |
OH |
||||
|
H |
C |
OH |
|||
|
HO |
C |
H |
|||
|
H |
C |
OH |
|||
|
H |
C |
OH |
|||
|
CH2OH |
CH2OH |
CH2OH |
|||
|
HOCH O |
HOCH O OH |
||||
|
OH |
OH |
||||
|
OH |
|||||
|
OH |
OH |
Существование таутомерных форм затрудняет определение состава моносахаридов в гидролизате.
Моносахариды нелетучи и непосредственное разделение их смесей методом газожидкостной хроматографии невозможно. Сахара могут быть хроматографически разделены в виде летучих производных — простых и сложных эфиров. Широко распространен метод анализа в виде триметилсилильных (ТМС) производных моносахаридов. Последние являются летучими гликозидами простых О-триметилсилиловых эфиров углеводов, образующихся в результате полного замещения гидроксильных групп моносахаридов при их силировании.
В качестве силирующего реагента моносахаридов используют смесь триметилхлорсилана и гексаметилдисилазана в среде пиридина. В качестве примера приводится схема реакции силирования для β —D- глюкозы:
|
CH2OH |
[(CH3)3Si]2NH |
CH2OSi(CH3)3 |
||
|
O OH |
(CH3)3SiCl |
(CH3)3Si |
O OSi(CH3)3 |
|
|
OH |
O |
|||
|
пиридин |
||||
|
HO |
(CH3)3SiO |
|||
|
OH |
OSi(CH3)3 |
138
Подобным образом синтезируются ТМС-производные других моносахаридов. При этом устанавливается новая равновесная смесь, в которой преобладают пиранозные формы. Соотношение таутомеров в зависимости от условий колеблется. Состав ТМС-производных таутомерных форм индивидуальных моносахаридов приведен ниже.
|
Таутомерные формы моносахаридов |
Массовая доля, % |
|
L — A р а б и н о з а |
100 |
|
α -Арабинопираноза |
51,6 |
|
β -Арабинопираноза |
40,1 |
|
Арабинофураноза |
8,3 |
|
D — К с и л о з а |
100 |
|
α -Ксилопираноза |
40,2 |
|
β -Ксилопираноза |
56,2 |
|
Ксилофураноза |
3,6 |
|
D — М а н н о з а |
100 |
|
α -Маннопираноза |
72,8 |
|
β -Маннопираноза |
27,2 |
|
D — Г а л а к т о з а |
100 |
|
α -Галактопираноза |
29,9 |
|
β -Галактопираноза |
45,8 |
|
Галактофураноза |
24,3 |
|
D — Г л ю к о з а |
100 |
|
α -Глюкопираноза |
47,3 |
|
β -Глюкопираноза |
49,5 |
|
Глюкофураноза |
3,2 |
Для уменьшения числа пиков на хроматограмме и упрощения ее обработки моносахариды восстанавливают до соответствующих многоатомных спиртов (полиолов) с последующим их превращением в ацетаты. Каждый полиол даст на хроматограмме один пик, поскольку таутомерия у полиолов невозможна.
При анализе ТМС-производных методом газожидкостной хроматографии используют в качестве газа-носителя азот или гелий. В качестве жидкой фазы используют различные силиконовые масла и полиэфиры. Наиболее селективными фазами из вырабатываемых отечественной промышленностью являются неполярное метилхлорсиликоновое масло и полярный политриэтиленгликольсебацинат. В качестве твердого носителя применяют адсорбенты порохром-3 или хромат N (за рубежом широко применяют хромосорб-W).

139
Рисунок 35 − Хроматограмма модельной смеси ТМС производных моносахаридов:
1 − β —L-арабиноза; 2 − γ —D— ксилоза; 3 − α —L-арабиноза; 4 − γ —L— арабиноза; 5 − α -D-ксилоза; 6 − β —D— ксилоза; 7 − α —D-манноза; 8 − γ —D— галактоза; 9 − α —D-галактоза; 10 − β — D-манноза; 11 − α —D-глюкоза + β -D- галактоза; 12 − сорбит; 13 − β —D— глюкоза
Для хроматографического разделения смеси летучих производных моносахаридов используют ДИП. В соответствии с tR на хроматограмме в определенном порядке появляются пики индивидуальных соединений. Порядок выхода при хроматографическом разделении ТМС-производных зависит от жидкой фазы. Его устанавливают предварительно для данной фазы с использованием модельных ТМС-производных чистых моносахаридов по их времени удерживания. В качестве примера на рисунке 35 приведена хроматограмма модельной смеси, разделенной на колонке с метилхлорфенилсиликоновым маслом в качестве жидкой фазы.
Характеристики удерживания ТМС-производных сахаров при хроматографии в следующих условиях: колонка из стали; длина − 3 м; диаметр − 3 мм; твердый носитель − хромосорб (120/140 меш); НЖФ − полиметилфенилсиликоновая жидкость (НМФМ-6); температура колонки − 170° С; изотермический режим; газ-носитель − гелий; расход газа − 60
|
мл/мин; температура испарения − |
250° С приведены в таблице 13. |
|||
|
Таблица 13 − Время удерживания ТМС-производных tR |
||||
|
Компонент |
tR |
Компонент |
tR |
|
|
β —L-Арабиноза |
7′48″ |
γ —D-Галактоза |
20′54″ |
|
|
α —D-Ксилоза |
8′48″ |
α —D-Галактоза |
23′00″ |
|
|
α —L-Арабиноза |
9′24″ |
β —D-Манноза |
25′42″ |
|
|
γ —D-Арабиноза |
10′42″ |
α —D-Глюкоза |
27′24″ |
|
|
γ —D-Ксилоза |
12′30″ |
β —D-Галактоза |
28′12″ |
|
|
β —D-Ксилоза |
15′30″ |
Сорбит |
33′30″ |
|
|
α —D-Манноза |
17′30″ |
β —D-Глюкоза |
40′30″ |
Существует несколько методов количественного определения состава смеси по полученным хроматограммам. По первому методу опре-
140
деляют площади пиков всех таутомеров каждого моносахарида расчетом площади треугольной аппроксимацией пика — умножением основания пика на половину высоты. При частичном наложении пиков друг на друга и невозможности прямого измерения основания площадь пика определяют умножением его высоты на ширину, измеренную на половине высоты. Возможно также определять долю каждого пика его переносом на бумагу и взвешиванием вырезанного из нее пика на аналитических весах, относя найденную массу к массе всех анализируемых пиков. Результат выражают в процентах, используя метод нормировки площадей.
По второму методу количественное определение массовой доли конкретного моносахарида в смеси производят по площади пика одного из его таутомеров, исходя из условия, что при синтезе ТМС-производных в строго постоянных условиях соотношение таутомеров сохраняется постоянным. Это позволяет для расчета выбрать наиболее разрешенный пик, называемый характеристическим. В качестве таких пиков обычно используют пики ТМС-производных β —L-арабинопиранозы, β —D-кси- лопиранозы, α —D-маннопиранозы, α —D-галактопиранозы и β -D- глюкопиранозы. Площади пиков остальных таутомеров непосредственно не измеряют, а находят расчетом. С учетом доли характеристического пика вычисляют общую площадь пиков всех таутомеров данного моносахарида. Количественное соотношение таутомерных форм необходимо устанавливать для данных условий хроматографирования и при их изменении определение следуют проводить вновь. Долю конкретного моносахарида находят отнесением площади всех пиков данного моносахарида к общей площади всех пиков.
Количественное определение массы индивидуальных моносахаридов производят с использованием внутреннего стандарта (сорбита). Масса сорбита, добавляемая в анализируемую смесь, должна обеспечить его концентрацию в растворе, примерно соответствующую концентрации индивидуальных моносахаридов. Расчет ведут по градуировочному графику. График получают, по данным хроматографирования эталонных растворов, содержащих индивидуальный моносахарид и сорбит. Градуировочный график строят в координатах: по оси абсцисс — масса чистого моносахарида в эталонном растворе, по оси ординат — отношение площади характеристического пика этого же моносахарида к площади пика внутреннего стандарта. График имеет линейную форму. Прямую линию проводят с использованием метода наименьших квадратов.
Относительная ошибка определений сахаров в гидролизате зависит от их массовой доли в анализируемой смеси и составляет 1− 5%. Общее время анализа при серийном проведении определений около 2 ч, не считая времени на получение градуировочных графиков.
Соседние файлы в предмете Химия
- #
16.08.201311.96 Mб29Халецкий А.М. Фармацевтическая химия. 1966.djvu
- #
- #
16.08.20139.41 Mб37Хмельницкий Л.И. Справочник по взрывчатым веществам (часть 2). 1962.djvu
- #
- #
- #
- #
- #
- #
- #
- #
Площадь — пик — хроматограмма
Cтраница 1
Площадь пиков хроматограммы является основой для количественных расчетов концентраций компонентов. Сумму площадей пиков принимают за 100 % и рассчитывают содержание отдельного компонента отношением площадей пика компонента к общей площади пиков. Этот метод ( рис. 50) может быть использован для расчета массовых процентов при анализе близкокипящих соединений, соответствующих одному гомологическому ряду. Площадь пика чаще всего определяется как площадь треугольника.
[1]
Поэтому по площади пиков хроматограмм рассчитывают концентрации компонентов анализируемой смеси.
[2]
Разработано устройство ЦИУ-2 для автоматического интегрирования площади пиков хроматограмм с высокой степенью точности и выдающее результаты в цифровой форме, удобной для связи хроматографа с ЦВМ. Приведены блок-схема устройства и принципиальные электрические схемы его отдельных элементов.
[3]
Ниже рассмотрены методы, которые используют для определения площадей пиков хроматограммы.
[5]
Для автоматической обработки информации газовой хроматографии при определении площадей пиков хроматограмм первоначально использовали дисковые интеграторы. Это были механические приборы и достигаемая с их помощью точность по сравнению с вычислениями вручную была достаточно высока.
[6]
Число импульсов п, полученное в блоке частоты 7, пропорционально площади пика хроматограммы, передается в регистр, связанный со схемой цифровой индикации на декатронах, и может быть зафиксировано циф-ропечатающей машиной.
[7]
Шнейдер [174] описали новый тип интегратора для газовой хроматографии, позволяющий очень точно определять площади пиков хроматограммы с учетом чувствительности и других факторов. Все это делается автоматически с использованием нового принципа устройства интегратора.
[8]
При проверке возможности использования исследованных неподвижных фаз для количественного определения фурана и его гомологов в их смесях были приготовлены модельные смеси, содержащие до 20 % фурана, метилфурана, этилфурана, пропилфурана и бутилфура-на, а также смеси, содержание по 20 % фурана, метилфурана, пропилфурана и 40 % бутилфурана. По площадям пиков хроматограмм, полученных при температурах 100, 120 и 150 С, был рассчитан состав смесей. Оказалось, что максимальная ошибка составляет 1 5 абс.
[10]
Пользуясь интегратором или находя произведение высоты каждого пика на его ширину, измеренную на половине высоты, находят площади пиков хромато-граммы, относящиеся к каждому интервалу времени. Принимая всю площадь пиков хроматограммы за 100 %, рассчитывают массу дистиллированной части в процентах в конце каждого выбранного интервала времени и строят кривую: масса дистиллированной части в процентах-время удерживания. Пользуясь этой кривой и кривой температур кипения нормальных парафинов, интерполируют значения содержания углеводородов с различными температурами кипения в пробе.
[11]
Предлагается использовать отношение площадей пиков хроматограмм продуктов пиролиза стандартного сополимера стирола и изопрена к площади пика дипенте-на для определения температуры пиролиза в пиролизере с металлической лентой.
[12]
Подготовленную к анализу ячейку соединяют вакуумной резиновой трубкой со штуцером крана-дозатора хроматографа. Концентрацию кислорода определяют по площади пика хроматограммы.
[14]
После выполнения описанной подготовки приступают к ана лизу. Естественно, что, во-первых, сумма площадей пиков хроматограммы, полученной при разделении битума, соответствует 100 %, а сумма площадей пиков хроматрграммы, полученной при разделении мальтенов, меньше 100 % на величину, соответствующую содержанию асфальтенов; во-вторых, площадь пика на хроматограмме мальтенов, обязанного спирто-бензолу, меньше площади аналогичного пика на хроматограмме битума на величину, соответствующую содержанию асфальтенов. Это позволяет рассчитать содержание в битуме мальтенов и асфальтенов — разумеется, при одинаковом количестве взятых на анализ мальтенов и битума; однако разность этих количеств легко учитывается сравнением площадей пиков, обязанных изооктану, на хроматограммах битума и мальтенов.
[15]
Страницы:
1
2
Процесс обработки хроматографического сигнала состоит из нескольких этапов: фильтрация шумов, разметка пиков, идентификация пиков, градуировка, количественный расчет. В этой статье мы рассмотрим один из наиболее важных вопросов — разметку пиков и их основные параметры. Мы расскажем как происходит разметка пиков на примере программы NetChrom, разработанной компанией «Мета-хром».
- Главные параметры пиков
- Типы хроматографических пиков
- Автоматическая разметка пиков
- Ручная разметка пиков
- Идентификация пиков:
- по абсолютному времени удерживания
- по относительному времени удерживания
- по относительному объему удерживания
- по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах
- по индексам удерживания (Ковача)
- по температурам кипения
Главные параметры пиков
Разметка пиков (интегрирование) — операция вычисления параметров пиков, полученных на хроматограмме путем определения характерных точек (начало, вершина и конец пика). При этом пики ограничиваются базовой линией (прямой, соединяющей точки начала и конца пика на нулевой линии), которая проводится по методу «резиновой ленты», натягивающейся снизу от точки начала до точки конца базовой линии на протяжении всей хроматограммы.
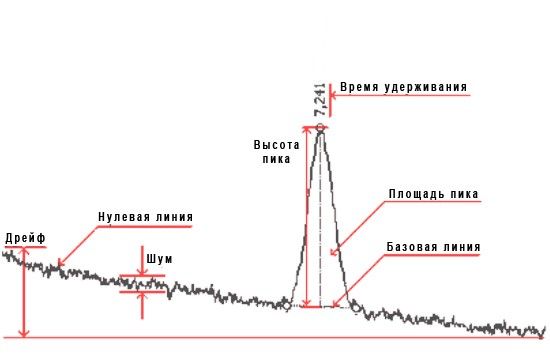
На основании вычислений оцениваются основные параметры пика:
- время удерживания — время от начала анализа до максимума пика;
- площадь — область, заключенная между пиком и ограничивающей его базовой линией;
- ысота — расстояние между базовой линией и максимумом пика;
- ширина пика на половине его высоты.
Важно! Особенностью программы NetChrom является полностью автоматизированный процесс ее настройки под конкретный хроматографический сигнал, непрерывное отслеживание изменения характеристик хроматографического сигнала в ходе обработки хроматограммы: уровня шумов и дрейфа, а также уровня самого сигнала.
Для определения характерных точек пиков: начала, конца и вершины — в программе NetChrom используется алгоритм на основе вычисления первой сглаженной производной хроматографического сигнала, скорректированной на дрейф базовой линии.
При превышении или достижении определенного уровня этой производной, называемой порогом, определяются характерные точки пиков. Уровень порога определяется на основе шума производной на участках хроматографического сигнала, свободного от пиков.
Кроме уровня шума, дрейфа и уровня сигнала, большое значение имеет также зависимость изменения ширины хроматографических пиков от времени их удерживания в процессе анализа (ширина пика в начале и в конце анализа).
Как известно, ширина хроматографических пиков непрерывно увеличивается в процессе анализа, причем в изотермическом режиме эта зависимость имеет линейный характер, в режиме программирования температуры колонок эта зависимость имеет более сложный характер. Программа использует эту зависимость для автоматической настройки программных фильтров под хроматографический сигнал во время анализа.
Кроме этого, пользователю предоставлена возможность сформировать или отредактировать зависимость изменения ширины хроматографических пиков от времени удерживания самостоятельно (ширина пиков в начале и в конце хроматограммы).
При неправильном задании этой зависимости возможно некорректное определение характерных точек пика и даже пропуск небольших пиков. Зависимость изменения ширины хроматографических пиков от времени индивидуальна для каждого метода, и после изменения условий анализа: температуры колонок, расхода газоносителя и др., необходимо откорректировать зависимость.
Типы хроматографических пиков
Пики могут быть нескольких типов:
- простые пики — начало и конец пика принадлежат базовой линии;
- хвостатые пики — асимметрия заднего фронта пика;
- пики наездники — пики на хвосте большего по величине пика;
- неразделенные пики — конец первого пика совпадает с началом второго и эта точка не принадлежит базовой линии;
- зашкаленные пики — пики с плоской вершиной.
Разделение слившихся пиков производится по перпендикуляру или тангенте в зависимости от соотношения ширины и высоты этих пиков.
Пики наездники отделяются по тангенте и площадь под ними включается в площадь хвостатого пика.
Следует иметь в виду, что никакой алгоритм не может в ряде случаев гарантировать корректную разметку на пики, поскольку само понятие «пик» во многом субъективно и зависит от конкретной аналитической задачи. Например, нельзя гарантировать корректную разметку на пики при сложной форме базовой линии, плохом разделении хроматографических пиков, малых пиках-наездниках, высоком уровне шумов и так далее.
При этом правильность получаемых результатов зачастую зависит от опыта пользователя. В программе NetChrom реализованы два подхода для проведения разметки пиков:
- автоматическая разметка пиков;
- ручная (графическая) разметка пиков.
Ниже мы рассмотрим каждый из подходов более детально.
Автоматическая разметка пиков
Автоматическая разметка пиков (интегрирование) имеет смысл тогда, когда ожидается обработка серии хроматограмм со сходными, повторяющимися особенностями базовой линии, значениями величины и последовательностью хроматографических пиков.
Параметры интегрирования, с помощью которых пользователь может влиять на процесс обнаружения пиков на хроматограмме, представлены ниже:
Ширина

Пороги
Порог обнаружения пиков может быть задан по двум параметрам: минимальной площади и минимальной высоте пика.
- Минимальная площадь
Минимально допустимая площадь детектируемого пика. При детектировании пиков имеется возможность не размечать или подавлять пики, площадь которых меньше заданной. При этом значение параметра, равное 0, означает, что подавление пиков выключено.

- Минимальная высота
Минимально допустимая высота детектируемого пика. Подавление пиков с высотой значение которой, меньше заданного. При этом значение параметра, равное 0, означает, что подавление пиков выключено.

Не всегда получается подобрать одинаковые параметры для разметки всех пиков. Например, с увеличением времени выхода пика, изменяется его ширина, поэтому значения параметров, подходящие для пиков в начале хроматограммы, могут не подходить для пиков в ее конце. В таких случаях рекомендуется использовать события интегрирования.
Настройка алгоритма разметки с использованием событий интегрирования имеет смысл, если ожидается серия однотипных хроматограмм со сходными, повторяющимися особенностями базовой линии. События интегрирования позволяют настроить процесс разметки в соответствии с особенностями данной серии хроматограмм, задавая для некоторых участков хроматограмм индивидуальные параметры и правила разметки.
Событие можно задать для отдельного детектора, и оно начинает действовать с указанного момента времени до тех пор, пока не будет переопределено другим событием такого же типа, или пока не завершится хроматограмма. Если не удается добиться желаемой разметки при использовании параметров и событий интегрирования, используют ручное графическое интегрирование (непосредственно на графике хроматограммы).
Ручная разметка пиков
Ручная разметка пиков используется, если не удается добиться желаемой разметки при использовании параметров автоматической разметки. Ручная графическая разметка пиков производится непосредственно на графике хроматограммы. При этом все действия, выполняемые пользователем, относятся к выделенному фрагменту хроматограммы или к выделенному пику. В программе существуют следующие типовые операции по ручному редактированию пиков на хроматограмме:
- создание пика;
- разделение пика;
- слияние пиков;
- корректировка положения характерных точек пика;
- задание пика наездником;
- задание пика хвостатым;
- задание пика базовым;
- задание пика слившимся;
- удаление пика;
- удаление пиков справа;
- удаление пиков слева;
- удаление всех пиков.
Идентификация пиков
Идентификация — отнесение пиков на хроматограмме к тому или иному компоненту из списка компонентов рабочего метода. При этом производится сравнение рассчитанных параметров удерживания всех обнаруженных на хроматограмме пиков с информацией, хранящейся в таблице компонентов. Идентификация компонентов по одному или по нескольким детекторам осуществляется следующими способами:
- по абсолютному времени удерживания;
- по относительному времени удерживания;
- по относительному объему удерживания;
- по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах;
- по индексам удерживания (Ковача);
- по температурам кипения.
Теперь рассмотрим каждый из вышеперечисленных способов подробнее.
Идентификация по абсолютному времени удерживания
Наиболее простой способ идентификации — по времени удерживания, то есть сравнение времени удерживания анализируемого компонента со временем удерживания известного соединения при строго заданных условиях анализа. Для проведения идентификации пика по времени удерживания в библиотеке компонентов должна содержаться информация:
- наименование компонента;
- время удерживания;
- окно поиска по времени (в единицах времени).
Окно поиска — границы области, в которой будет осуществляться поиск пика как в положительную, так и в отрицательную сторону от заданного в таблице параметра удерживания.
При задании окна необходимо стремиться, чтобы его ширина была достаточной для попадания пика в окно при неизбежных изменениях времени удерживания, но и не слишком большой, чтобы в него не попадали соседние пики.
В случае если в окно поиска попадают несколько пиков, то среди них выбирается пик, имеющий максимальную вероятность идентификации (наиболее интенсивный или ближайший к библиотечному времени).
Идентификация по относительному времени удерживания
При изменении условий в процессе анализа (расход газа-носителя, температура колонки), а также в процессе «старения» колонок, пики могут не попасть в окно поиска. Это происходит чаще всего:
- когда нельзя задать достаточно широкое окно поиска, чтобы в него не попали соседние пики;
- при длительных анализах с программированием температуры колонок, когда время удерживания может измениться в большей степени.
Выход из положения состоит в том, что один из пиков (или несколько) назначается «стандартом времени», для него задается увеличенное окно поиска (2-5%), а для остальных пиков рассчитывается относительное время удерживания.
Стандартом времени, как правило, выбираются стоящие отдельно или большие пики обязательно присутствующие на хроматограмме. Таким образом, при одновременном сдвиге по той или иной причине времен удерживания всех компонентов, наличие пиков-стандартов времени поможет правильно идентифицировать вещества, несмотря на то, что их время удерживания не будет попадать в окно поиска по времени.
В этом случае идентификация производится следующим образом:
- производится поиск пика стандарта времени по времени удерживания;
- для стандарта времени удерживания рассчитывается коэффициент отклонения реального времени удерживания по сравнению с библиотечным временем по формуле:
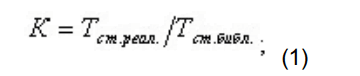
для остальных пиков рассчитывается ожидаемое время удерживания, исходя из времени, заданного в библиотеке для данного компонента, и рассчитанного коэффициента отклонения времени стандарта по формуле:
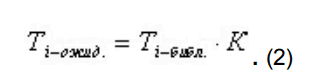
Идентификация по относительному объему удерживания
Является более точным расчетным параметром по сравнению с относительным временем удерживания, так как в нем учитывается время на прохождение подвижной фазой расстояния от устройства для ввода пробы до детектора (иногда это время называют «мертвым» временем или временем удерживания несорбирующегося вещества).
При этом относительный объем удерживания стандарта времени принимают за единицу, а относительный объем удерживания компонентов (Ri) рассчитывают по формуле:
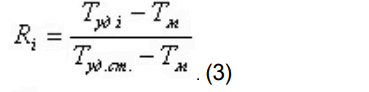
T уд i — время удерживания анализируемого компонента;
T уд.ст. — время удерживания стандарта времени;
T м — мертвое время.
Идентификация по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах
Если в процессе анализа используются несколько (чаще всего два) параллельно или последовательно работающих детектора, то для более достоверной идентификации можно применить способ идентификации, заключающийся в том, что наряду со временем удерживания (абсолютным или относительным), можно использовать отношение интенсивностей пиков соответствующих детекторов.
Вначале пик идентифицируется по времени удерживания на ведущем детекторе (детекторе, по интенсивности пика которого рассчитывается концентрация компонента). Затем сравниваются отношения интенсивностей пика на различных детекторах с библиотечным отношением.
Для идентификации может использоваться не только отношение интенсивностей, но также наличие или отсутствие пика на другом (не ведущем) детекторе.
Идентификация по индексам удерживания (Ковача)
Для идентификации могут использоваться и другие относительные параметры удерживания, которые в меньшей степени зависят (в отличие от времени удерживания) от условий анализа. Одним из таких параметров является индекс удерживания — безразмерная величина, характеризующая положение пика вещества на хроматограмме относительно пиков выбранных стандартов.
Если в качестве стандартов используются насыщенные углеводороды (алканы, парафины), то индекс удерживания называется индексом Ковача. Выбор типа индекса (линейный или логарифмический) зависит от условий анализа.
Для постоянной температуры колонки во время анализа характерна логарифмическая зависимость, при программировании — линейная. Однако между этими двумя зависимостями нет четкого разделения, поэтому при применении режима программирования температуры колонок с начальным изотермическим участком, используется смешанный тип индекса. При этом на изотермическом участке выбирается логарифмический тип индекса, до первого реперного пика, который попадает на участок программирования температур, и в дальнейшем — линейный тип индекса.
Начало программирования температуры колонок выбирается согласно заданным параметрам управления соответствующего метода. При идентификации по индексам удерживания в таблицу компонентов должны быть занесены табличные значения индекса компонентов и ширина окна поиска по индексу. Пикам – стандартам индексов удерживания необходимо присвоить тип: реперный в таблице компонентов и задать увеличенное окно поиска (2-5%) от времени удерживания.
Идентификация по индексам удерживания производится следующим образом:
- Производится идентификация реперных пиков по времени удерживания.
- Реперным пикам присваиваются соответствующие индексы из таблицы компонентов.
- Используя индексы удерживания реперных пиков, рассчитываются индексы удерживания обычных пиков и сравниваются с табличными данными.
Индексы удерживания Ковача рассчитываются по формулам:
линейный индекс удерживания:
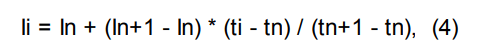
логарифмический индекс удерживания:
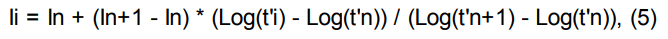
где:
Ii — время удерживания интересующего пика;
In, In+1 — индексы предыдущего и последующего компонентов с известной величиной индекса;
ti — время удерживания интересующего пика;
tn, tn+1 — времена удерживания пиков, соответствующие предыдущему и последующему компонентам с известными индексами;
t’… — приведенное время удерживания.
Важно! Для увеличения точности расчета индексов удерживания в качестве времени удерживания необходимо брать приведенное время удерживания. Приведенное время удержания равно разности абсолютного времени удержания и мертвого времени (времени нахождения неудерживаемого компонента в хроматографической системе).
Мертвое время определяется либо экспериментально, либо расчетом, выполненным с помощью газового калькулятора. В качестве неудерживаемого компонента чаще всего используют метан.
В окно ожидаемого времени или индекса удерживания одного компонента может попасть несколько пиков. В этом случае выбор пика, в зависимости от настройки, может осуществляться по следующим критериям:
- ближайший к ожидаемому (библиотечному) времени или индексу удерживания;
- максимальный по высоте;
- максимальный по площади.
Один и тот же пик может попасть в окна поиска разных компонентов, в этом случае необходимо уменьшить ширину окон поиска.
Указанная схема распознавания (идентификации) компонентов оказывается достаточно универсальной и гибкой для того, чтобы проблема корректного распознавания пиков в подавляющем большинстве случаев было решена. Все, что требуется от оператора — на этапе настройки грамотно выбрать пик стандарта времени или реперные пики и в дальнейшем периодически корректировать их ожидаемые времена удерживания по текущей хроматограмме.
Идентификация по температурам кипения
Частным случаем расчета индексов удерживания является расчет температур кипения. При расчете температур кипения используются те же формулы, что и при расчете индексов. Для расчета температур кипения необходимо в библиотеке методов выбрать тип индекса (линейный или логарифмический) и задать известные температуры кипения реперных компонентов.