From Wikipedia, the free encyclopedia
«LOQ» redirects here. For the company listed as LOQ on the London Stock Exchange, see Lo-Q. For the airport in Botswana with IATA code LOQ, see Lobatse Airport.
The limit of detection (LOD or LoD) is the lowest signal, or the lowest corresponding quantity to be determined (or extracted) from the signal, that can be observed with a sufficient degree of confidence or statistical significance. However, the exact threshold (level of decision) used to decide when a signal significantly emerges above the continuously fluctuating background noise remains arbitrary and is a matter of policy and often of debate among scientists, statisticians and regulators depending on the stakes in different fields.
Significance in analytical chemistry[edit]
In analytical chemistry, the detection limit, lower limit of detection, also termed LOD for limit of detection or analytical sensitivity (not to be confused with statistical sensitivity), is the lowest quantity of a substance that can be distinguished from the absence of that substance (a blank value) with a stated confidence level (generally 99%).[1][2][3] The detection limit is estimated from the mean of the blank, the standard deviation of the blank, the slope (analytical sensitivity) of the calibration plot and a defined confidence factor (e.g. 3.2 being the most accepted value for this arbitrary value).[4] Another consideration that affects the detection limit is the adequacy and the accuracy of the model used to predict concentration from the raw analytical signal.[5]
As a typical example, from a calibration plot following a linear equation taken here as the simplest possible model:

where,  corresponds to the signal measured (e.g. voltage, luminescence, energy, etc.), «b» the value in which the straight line cuts the ordinates axis, «a» the sensitivity of the system (i.e., the slope of the line, or the function relating the measured signal to the quantity to be determined) and «x» the value of the quantity (e.g. temperature, concentration, pH, etc.) to be determined from the signal ,[6] the LOD for «x» is calculated as the «x» value in which equals to the average value of blanks «y» plus «t» times its standard deviation «s» (or, if zero, the standard deviation corresponding to the lowest value measured) where «t» is the chosen confidence value (e.g. for a confidence of 95% it can be considered t = 3.2, determined from the limit of blank).[4]
corresponds to the signal measured (e.g. voltage, luminescence, energy, etc.), «b» the value in which the straight line cuts the ordinates axis, «a» the sensitivity of the system (i.e., the slope of the line, or the function relating the measured signal to the quantity to be determined) and «x» the value of the quantity (e.g. temperature, concentration, pH, etc.) to be determined from the signal ,[6] the LOD for «x» is calculated as the «x» value in which equals to the average value of blanks «y» plus «t» times its standard deviation «s» (or, if zero, the standard deviation corresponding to the lowest value measured) where «t» is the chosen confidence value (e.g. for a confidence of 95% it can be considered t = 3.2, determined from the limit of blank).[4]
Thus, in this didactic example:

There are a number of concepts derived from the detection limit that are commonly used. These include the instrument detection limit (IDL), the method detection limit (MDL), the practical quantitation limit (PQL), and the limit of quantitation (LOQ). Even when the same terminology is used, there can be differences in the LOD according to nuances of what definition is used and what type of noise contributes to the measurement and calibration.[7]
The figure below illustrates the relationship between the blank, the limit of detection (LOD), and the limit of quantitation (LOQ) by showing the probability density function for normally distributed measurements at the blank, at the LOD defined as 3 × standard deviation of the blank, and at the LOQ defined as 10 × standard deviation of the blank. For a signal at the LOD, the alpha error (probability of false positive) is small (1%). However, the beta error (probability of a false negative) is 50% for a sample that has a concentration at the LOD (red line). This means a sample could contain an impurity at the LOD, but there is a 50% chance that a measurement would give a result less than the LOD. At the LOQ (blue line), there is minimal chance of a false negative.
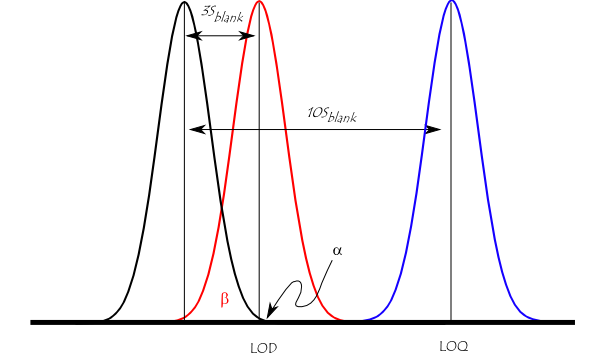
Illustration of the concept of detection limit and quantitation limit by showing the theoretical normal distributions associated with blank, detection limit (LOD), and quantitation limit (LOQ) level samples.
Instrument detection limit[edit]
Most analytical instruments produce a signal even when a blank (matrix without analyte) is analyzed. This signal is referred to as the noise level. The IDL is the analyte concentration that is required to produce a signal greater than three times the standard deviation of the noise level. This may be practically measured by analyzing 8 or more standards at the estimated IDL then calculating the standard deviation from the measured concentrations of those standards.
The detection limit (according to IUPAC) is the smallest concentration, or the smallest absolute amount, of analyte that has a signal statistically significantly larger than the signal arising from the repeated measurements of a reagent blank.
Mathematically, the analyte’s signal at the detection limit ( ) is given by:
) is given by:

where,  is the mean value of the signal for a reagent blank measured multiple times, and
is the mean value of the signal for a reagent blank measured multiple times, and  is the known standard deviation for the reagent blank’s signal.
is the known standard deviation for the reagent blank’s signal.
Other approaches for defining the detection limit have also been developed. In atomic absorption spectrometry usually the detection limit is determined for a certain element by analyzing a diluted solution of this element and recording the corresponding absorbance at a given wavelength. The measurement is repeated 10 times. The 3σ of the recorded absorbance signal can be considered as the detection limit for the specific element under the experimental conditions: selected wavelength, type of flame or graphite oven, chemical matrix, presence of interfering substances, instrument… .
Method detection limit[edit]
Often there is more to the analytical method than just performing a reaction or submitting the analyte to direct analysis. Many analytical methods developed in the laboratory, especially these involving the use of a delicate scientific instrument, require a sample preparation, or a pretreatment of the samples prior to being analysed. For example, it might be necessary to heat a sample that is to be analyzed for a particular metal with the addition of acid first (digestion process). The sample may also be diluted or concentrated prior to analysis by means of a given instrument. Additional steps in an analysis method add additional opportunities for errors. Since detection limits are defined in terms of errors, this will naturally increase the measured detection limit. This «global» detection limit (including all the steps of the analysis method) is called the method detection limit (MDL). The practical way for determining the MDL is to analyze seven samples of concentration near the expected limit of detection. The standard deviation is then determined. The one-sided Student’s t-distribution is determined and multiplied versus the determined standard deviation. For seven samples (with six degrees of freedom) the t value for a 99% confidence level is 3.14. Rather than performing the complete analysis of seven identical samples, if the Instrument Detection Limit is known, the MDL may be estimated by multiplying the Instrument Detection Limit, or Lower Level of Detection, by the dilution prior to analyzing the sample solution with the instrument. This estimation, however, ignores any uncertainty that arises from performing the sample preparation and will therefore probably underestimate the true MDL.
Limit of each model[edit]
The problem of limit of detection, or limit of quantification, is encountered in all scientific disciplines. This explains the variety of definitions and the diversity of solutions developed to address the question. In the simplest cases as in nuclear and chemical measurements, definitions and approaches have probably received the clearer and the simplest solutions. In biochemical tests and in biological experiments depending on many more intricate factors, the situation involving false positive and false negative responses is more delicate to handle. In many other disciplines such as geochemistry, seismology, astronomy, dendrochronology, climatology, life sciences in general, and in many other fields impossible to enumerate extensively, the problem is wider and deals with signal extraction out of a background of noise. It involves complex statistical analysis procedures and therefore it also depends on the models used,[5] the hypotheses and the simplifications or approximations to be made to handle and manage uncertainties. When the data resolution is poor and different signals overlap, different deconvolution procedures are applied to extract parameters. The use of different phenomenological, mathematical and statistical models may also complicate the exact mathematical definition of limit of detection and how it is calculated. This explains why it is difficult to find a general consensus about the precise mathematical definition of the notion of limit of detection. However, one thing is clear: it always requires a sufficient number of data (or accumulated data) and a rigorous statistical analysis to be statistically significant.
Limit of quantification[edit]
The limit of quantification (LoQ, or LOQ) is the lowest value of a signal (or concentration, activity, response…) that can be quantified with acceptable precision and accuracy.
The LoQ is the limit at which the difference between two distinct signals / values can be discerned with a reasonable certainty, i.e., when the signal is statistically different from the background. The LoQ may be drastically different between laboratories, so another detection limit is commonly used that is referred to as the Practical Quantification Limit (PQL).
See also[edit]
- Background noise – Sound other than the sound being monitored (primary sound)
- Background radiation – Measure of ionizing radiation in the environment
- Electronic noise – Random fluctuation in an electrical signal
- Noise (spectral phenomenon) – Types of noise
- Chemometrics – Science of extracting information from chemical systems by data-driven means
- Gamma spectroscopy#Calibration and background radiation – Quantitative study of the energy spectra of gamma-ray sources
- Malmquist bias – Sampling bias in astronomy
- p-value – Function of the observed sample results
- Misuse of p-values – Misinterpretation of statistical significance
- Statistical significance – Concept in inferential statistics
References[edit]
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the «Gold Book») (1997). Online corrected version: (2006–) «detection limit». doi:10.1351/goldbook.L03540
- ^ MacDougall D, Crummett WB, et al. (1980). «Guidelines for Data Acquisition and Data Quality Evaluation in Environmental Chemistry». Analytical Chemistry. 52 (14): 2242–49. doi:10.1021/ac50064a004.
- ^ Saah AJ, Hoover DR (1998). «[Sensitivity and specificity revisited: significance of the terms in analytic and diagnostic language]». Ann Dermatol Venereol. 125 (4): 291–4. PMID 9747274.
- ^ a b Armbruster DA, Pry T (August 2008). «Limit of blank, limit of detection and limit of quantitation». The Clinical Biochemist. Reviews. 29 Suppl 1 (1): S49–S52. PMC 2556583. PMID 18852857.
- ^ a b «R: «Detection» limit for each model». search.r-project.org. Retrieved 2022-01-04.
- ^ Quesada-González D, Stefani C, González I, de la Escosura-Muñiz A, Domingo N, Mutjé P, Merkoçi A (September 2019). «Signal enhancement on gold nanoparticle-based lateral flow tests using cellulose nanofibers». Biosensors & Bioelectronics. 141: 111407. doi:10.1016/j.bios.2019.111407. hdl:10261/201014. PMID 31207571. S2CID 190531742.
- ^ Long, Gary L.; Winefordner, J. D. (1983), «Limit of detection: a closer look at the IUPAC definition», Anal. Chem., 55 (7): 712A–724A, doi:10.1021/ac00258a724
Further reading[edit]
- Altshuler B, Pasternack B (1963-03-01). «Statistical measures of the lower limit of detection of a radioactivity counter». Health Physics. 9 (3): 293–298. doi:10.1097/00004032-196303000-00005. ISSN 0017-9078. PMID 14040764. Retrieved 2022-01-03.
- Currie LA (1968). «Limits for qualitative detection and quantitative determination. Application to radiochemistry». Analytical Chemistry. 40 (3): 586–593. doi:10.1021/ac60259a007. ISSN 0003-2700.
- Long GL, Winefordner JD (1983). «Limit of detection. A closer look at the IUPAC definition». Analytical Chemistry. 55 (7): 712A–724A. doi:10.1021/ac00258a001. ISSN 0003-2700.
- Armbruster DA, Pry T (August 2008). «Limit of blank, limit of detection and limit of quantitation». The Clinical Biochemist. Reviews. 29 Suppl 1 (Suppl 1): S49–S52. PMC 2556583. PMID 18852857.
- European Commission. Joint Research Centre. (2016). Guidance document on the estimation of LOD and LOQ for measurements in the field of contaminants in feed and food. Luxembourg: Publications Office. doi:10.2787/8931. ISBN 9789279617683. Retrieved 2022-01-03.
- «DIN 32645 – Chemical analysis – Decision limit, detection limit and determination limit under repeatability conditions – Terms, methods, evaluation. Technical standard. Deutsches Institut für Normung, Berlin (DIN 32645:2008-11) | Via Engineering360» (in German). Published by Beuth Verlag, a subsidiary of the DIN Group. doi:10.31030/1465413. Retrieved 2022-01-03.
External links[edit]
- Evans WC (21 February 2019). «Limit of Detection – Interactive Java applet to illustrate some basic ideas of the limit of detection problem». GeoGebra. Retrieved 2022-01-04.
- «The R Language». search.r-project.org. Retrieved 2022-01-04.
- Garrett RG (2013-11-01). «The ‘rgr’ package for the R Open Source statistical computing and graphics environment – a tool to support geochemical data interpretation». Geochemistry: Exploration, Environment, Analysis. 13 (4): 355–378. doi:10.1144/geochem2011-106. ISSN 1467-7873. S2CID 129059022. Retrieved 2022-01-04.
- «R: «Detection» limit for each model». search.r-project.org. Retrieved 2022-01-04.
- Deutsches Institut für Normung. «R: Calibration data from DIN 32645 (Package envalysis version 0.5.1)». search.r-project.org. Retrieved 2022-01-04.
- Downloads of articles (a.o. harmonization of concepts by ISO and IUPAC) and an extensive list of references
Каждый
инструментальный метод характеризуется
определенным уровнем шумов, связанным
со спецификой измерительного процесса.
Поэтому всегда существует предел
содержаний, ниже которого вещество
вообще не может быть надежно обнаружено.
Предел
обнаружения
Сmin,
P
–
наименьшее содержание, при котором по
данной методике можно обнаружить
присутствие компонента с заданной
доверительной вероятностью.
Предел
обнаружения может быть и задан и
минимальным аналитическим сигналом
ymin
,
который можно
уверенно
отличать от сигнала контрольного опыта
– yфон.
Статистическими
методами с применением
неравенства Чебышева доказано, что
количественно предел обнаружения можно
определить, пользуясь выражением
![]()
Где
sфон
стандартное
отклонение аналитического сигнала
фона; S-
коэффициент чувствительности (его
иногда называют просто «чувствительность»)
, он характеризует отклик аналитического
сигнала на содержание компонента.
Коэффициент чувствительности – это
значение первой производной градуировочной
функции при данном определении
концентрации. Для прямолинейных
градуировочных графиков – это тангенс
угла наклона:
![]()
![]()
(внимание:
не
спутайте
коэффициент
чувствительности S
со стандартным
отклонением s!)
Существуют
и другие способы расчета предела
обнаружения, но данное уравнение
используют чаще всего.
В
количественном химическом анализе
обычно приводят диапазон определяемых
содержаний или концентраций. Он означает
область значений определяемых содержаний
(концентраций), предусмотренную данной
методикой и ограниченную нижней и
верхней границами определяемых
концентраций.
Аналитика
чаще интересует нижняя граница
определяемых концентрации сн
или содержания mн
компонента,
определяемого по данной методике. За
нижнюю границу определяемых содержаний
обычно принимают то минимальное
количество или концентрацию, которые
можно определить с относительным
стандартным отклонением
![]() ..
..
Пример
В
растворе определяли массовую концентрацию
железа спектрофотометрическим методом,
измеряя оптические плотности растворов,
окрашенных в результате реакции
взаимодействия иона Fe3+
с сульфосалициловой кислотой. Для
построения градуировочной зависимости
были измерены оптические плотности
растворов с возрастающими (заданными)
концентрациями железа, обработанных
сульфосалициловой кислотой.
|
xi |
0,010 |
0,020 |
0,030 |
0,040 |
0,050 |
|
yl |
0,100 |
0,210 |
0,290 |
0,420 |
0,530 |
Оптические
плотности раствора сравнения (контрольного
опыта на реактивы, т.е без добавления
железа, (фон) составили 0,002; 0,000; 0,008;
0,006; 0,003.
Рассчитайте
предел обнаружения железа.
Решение
1)
В результате вычислений методом
наименьших квадратов (см. пример для
контрольного задания № 5) получены
значения для построения градуировочного
графика.
Вычисленные
значения для построения градуировочного
графика
|
xi |
0,010 |
0,020 |
0,030 |
0,040 |
0,050 |
|
yl |
0,096 |
0,203 |
0,31 |
0,417 |
0,524 |
2)
Вычисляем коэффициент чувствительности,
т.е.угловой коэффициент градуировочной
зависимости (S)
по данным таблицы.
![]()
![]()
3)
Вычисляем стандартное
отклонение фонового сигнала,
что составляет 0,0032
единиц оптической плотности.
4)
Предел обнаружения составит, мг/см3
![]()
К онтрольное
онтрольное
задание № 6
Определить
предел обнаружения железа в воде.
Исходные
данные:
значения оптической плотности фона
(раствора сравнения) при построении
градуировочного графика для определения
железа составили 0,003; 0,001; 0,007; 0,005;
0,006; 0,003; 0,001; 0,005. Значения оптических
плотностей, соответствующие концентрациям
железа в растворе представлены в таблице
контрольного задания № 5.
Рассчитайте
предел обнаружения железа в мг/см3
по
коэффициентам чувствительности S,
вычисленным на основании данных,
полученных для построения градуировочного
графика методом наименьших квадратов
при выполнении контрольного задания №
5;
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
27.04.201939.19 Кб314,15,17,27,38,41,43,44,46,47,51,52.docx
- #
- #
Валидация аналитических методик (ОФС.1.1.0012.15)
Государственная фармакопея 13 издание (ГФ XIII)
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
ОФС вводится впервые
Валидация аналитической методики – это экспериментальное доказательство того, что методика пригодна для решения предполагаемых задач.
Настоящая общая фармакопейная статья регламентирует характеристики аналитических методик, определяемые с целью их валидации, и соответствующие критерии пригодности валидируемых методик, предназначенных для контроля качества лекарственных средств: фармацевтических субстанций и лекарственных препаратов.
Валидации подлежат методики количественного определения, в том числе методики определения примесей и методики определения предела содержания. Методики проверки подлинности подвергаются валидации при необходимости подтвердить их специфичность.
При валидации проводится оценка аналитической методики по перечисленным ниже характеристикам, выбираемым с учетом типовых рекомендаций, приведенных в таблице:
- специфичности (specificity);
- пределу обнаружения (detection limit);
- пределу количественного определения (quantitation limit);
- аналитической области (range);
- линейности (linearity);
- правильности (trueness);
- прецизионности (precision);
- устойчивости (robustness).
Таблица 1. Характеристики методик, определяемые при валидации
| Наименование характеристики | Основные типы методик | ||||
| Испытание на подлинность | Посторонние примеси | Количественное определение | |||
| Количественные методики | Предел содержания | Основного действующего вещества, нормируемых компонентов | Действующего вещества в тесте «Растворение» | ||
| Специфичность**) | Да | Да | Да | Да | Да |
| Предел обнаружения | Нет | Нет | Да | Нет | Нет |
| Предел количественного определения | Нет | Да | Нет | Нет | Нет |
| Аналитическая область | Нет | Да | Нет | Да | Да |
| Линейность | Нет | Да | Нет | Да | Да |
| Правильность | Нет | Да | * | Да | Да |
| Прецизионность:
– повторяемость (сходимость) – промежуточная (внутрилабораторная) прецизионность |
Нет Нет |
Да Да |
Нет Нет |
Да Да |
Да Нет |
| Устойчивость | Нет | * | * | * | * |
* может определяться при необходимости;
** отсутствие специфичности одной аналитической методики может быть компенсировано использованием другой аналитической методики.
Ревалидацию (повторную валидацию) методик проводят при изменении:
- технологии получения объекта анализа;
- состава лекарственного средства (объекта анализа);
- ранее утвержденной методики анализа.
Специфичность
Специфичность – это способность аналитической методики однозначно оценивать определяемое вещество в присутствии сопутствующих компонентов.
Доказательство специфичности валидируемой методики обычно основывается на рассмотрении полученных с ее использованием данных анализа модельных смесей известного состава.
Специфичность валидируемой методики может быть доказана также соответствующей статистической обработкой результатов анализов реальных объектов, выполненных с ее использованием и, параллельно, с использованием другой, заведомо специфичной, методики (методики, специфичность которой доказана).
1.1. Для методик испытаний на подлинность
Валидируемая методика (или совокупность методик) должна обеспечивать достоверную информацию о присутствии данного действующего вещества в субстанции или лекарственной форме при наличии в ее составе предусмотренных рецептурой компонентов, что подлежит экспериментальному подтверждению.
Подлинность действующего вещества в фармацевтической субстанции или лекарственном препарате устанавливают в сравнении со стандартным образцом или по физико-химическим или химическим свойствам, не характерным для других компонентов.
1.2. Для методик количественного определения и испытания на примеси
Для валидируемой методики количественного определения и испытаний на примеси применяют одинаковые подходы − должна быть оценена ее специфичность в отношении определяемого вещества, т. е. должно быть экспериментально подтверждено, что присутствие сопутствующих компонентов не влияет непредусмотренным образом на результат анализа.
Допускается оценка специфичности валидируемой методики как путем анализа модельных смесей известного состава, содержащих определяемое вещество, так и путем сравнения результатов анализов реальных объектов, полученных одновременно с использованием валидируемой и другой, заведомо специфичной методики. Результаты соответствующих экспериментов должны быть статистически обработаны.
Недостаток специфичности испытания может быть компенсирован другим (другими) дополнительным испытанием.
При валидации методик, если это целесообразно, могут использоваться образцы лекарственных средств, подвергнутые, с целью накопления в них примесей, воздействию экстремальных условий (света, температуры, влажности) или химически модифицированные любым подходящим способом.
Для хроматографических методик показывают разрешение между двумя наиболее близко элюирующимися веществами при соответствующих концентрациях.
ПРЕДЕЛ ОБНАРУЖЕНИЯ
Предел обнаружения – это наименьшее количество (концентрация) определяемого вещества в образце, которое может быть обнаружено (или приближенно оценено) с использованием валидируемой методики.
Предел обнаружения в случаях, указанных в таблице, обычно выражается как концентрация определяемого вещества (в % относительных или долях на миллион – ppm).
В зависимости от типа методики (визуальная или инструментальная) используют разные способы определения предела обнаружения.
2.1. Для методик с визуальной оценкой результата анализа
Проводят испытания образцов с различными известными количествами (концентрациями) определяемого вещества и устанавливают минимальное значение, при котором результат анализа может быть оценен визуально. Это значение является оценкой предела обнаружения.
2.2. Для методик с инструментальной оценкой результата анализа
2.2.1. По соотношению сигнал/шум
Этот подход применим к методам, для которых наблюдается шум базовой линии. Сравнивают величины сигналов, полученных для контрольного опыта и для образцов с низкими концентрациями анализируемого вещества. Устанавливают минимальное количество (концентрацию) определяемого вещества в образце, при котором величина отношения аналитического сигнала к уровню шумов равна 3.
Найденная величина является оценкой предела обнаружения.
2.2.2. По величине стандартного отклонения сигнала и угловому коэффициенту калибровочного графика
Предел обнаружения (ПО) находят по уравнению:
ПО = 3,3 · S/b,
где S – стандартное отклонение аналитического сигнала;
b – коэффициент чувствительности, представляющий собой отношение аналитического сигнала к определяемой величине (тангенс угла наклона калибровочной кривой).
При наличии экспериментальных данных в широком диапазоне измеряемой величины S и b могут быть оценены методом наименьших квадратов.
Для линейного калибровочного графика значение S принимают равным стандартному отклонению Sa свободного члена уравнения этого графика. Полученное значение предела обнаружения при необходимости может быть подтверждено прямым экспериментом при количествах (концентрациях) определяемого вещества, близких к найденному значению предела обнаружения.
Как правило, если имеются данные о пригодности методики для надежного определения вещества в концентрациях, лежащих как выше, так и ниже нормы его содержания, установленной спецификацией, определять реальный предел обнаружения для такой методики не требуется.
ПРЕДЕЛ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ
Предел количественного определения – это наименьшее количество (концентрация) вещества в образце, которое может быть количественно оценено с использованием валидируемой методики с требуемой правильностью и внутрилабораторной (промежуточной) прецизионностью.
Предел количественного определения является необходимой валидационной характеристикой методик, используемых для оценки малых количеств (концентраций) веществ в образце и, в частности, для оценки содержания примесей.
В зависимости от типа методики используют следующие способы нахождения предела количественного определения.
3.1. Для методик с визуальной оценкой результата анализа
Проводят испытания образцов с различными известными количествами (концентрациями) анализируемого вещества и устанавливают минимальное значение, при котором результат анализа может быть получен визуально с требуемой правильностью и внутрилабораторной (промежуточной) прецизионностью.
3.2. Для методик с инструментальной оценкой результата анализа
3.2.1. По соотношению сигнал/шум
Устанавливают минимальную концентрацию определяемого вещества в образце, при которой величина отношения аналитического сигнала к уровню шума составляет около 10:1.
3.2.2. По величине стандартного отклонения сигнала и угловому коэффициенту калибровочного графика
Предел количественного определения (ПКО) рассчитывают по уравнению:
ПКО = 10 · S/b,
где S – стандартное отклонение аналитического сигнала;
b – коэффициент чувствительности, представляющий собой отношение аналитического сигнала к определяемой величине.
При наличии экспериментальных данных в широком диапазоне измеряемой величины S и b могут быть оценены методом наименьших квадратов.
Для линейного калибровочного графика значение S принимают равным стандартному отклонению Sa свободного члена уравнения этого графика. Полученное значение предела количественного определения при необходимости может быть подтверждено прямым экспериментом при количествах (концентрациях) определяемого вещества, близких к найденному значению предела количественного определения.
Если имеются данные о способности методики надежно определять анализируемое вещество в концентрации выше и ниже установленной в спецификации нормы его содержания, определять реальное значение предела количественного определения для такой методики, как правило, не требуется.
АНАЛИТИЧЕСКАЯ ОБЛАСТЬ МЕТОДИКИ
Аналитическая область методики – это интервал между верхним и нижним значением аналитических характеристик определяемого компонента в объекте анализа (его количества, концентрации, активности и т. п.). В этом интервале результаты, получаемые с использованием валидируемой методики, должны иметь приемлемый уровень правильности и внутрилабораторной (промежуточной) прецизионности.
К величине аналитической области методик предъявляются следующие требования:
- методики количественного определения должны быть применимы в интервале от 80 до 120 % от номинального значения определяемой аналитической характеристики;
- методики оценки однородности дозирования должны быть применимы в интервале от 70 до 130 % от номинальной дозы;
- методики количественного определения, используемые при проведении теста «Растворение», обычно должны быть применимы в пределах от 50 до 120 % от ожидаемой концентрации действующего вещества в среде растворения;
- методики испытаний на чистоту должны быть применимы в интервале от «Предела количественного определения» или «Предела обнаружения» до 120 % от допустимого содержания определяемой примеси.
Аналитическая область методики может быть установлена по диапазону экспериментальных данных, удовлетворяющих линейной модели.
ЛИНЕЙНОСТЬ
Линейность методики – это наличие линейной зависимости аналитического сигнала от концентрации или количества определяемого вещества в анализируемой пробе в пределах аналитической области методики.
При валидации методики ее линейность в аналитической области про-веряют экспериментально измерением аналитических сигналов для не менее чем 5 проб с различными количествами или концентрациями определяемого вещества. Экспериментальные данные обрабатывают методом наименьших квадратов с использованием линейной модели:
y = b · x + a, где
- х – количество или концентрация определяемого вещества;
- y – величина отклика;
- b – угловой коэффициент;
- a – свободный член (ОФС «Статистическая обработка результатов химического эксперимента»).
Должны быть рассчитаны и представлены величины b, a и коэффициент корреляции r. В большинстве случаев используют линейные зависимости, отвечающие условию 0,99, и только при анализе следовых количеств рассматривают линейные зависимости, для которых 0,9.
В отдельных случаях возможность линейной аппроксимации экспериментальных данных обеспечивается лишь после их математического преобразования (например, логарифмирования).
Для некоторых методик анализа, в основу которых в принципе не может быть положена линейная зависимость между экспериментальными данными, определение концентрации или количества вещества проводят с использованием нелинейных калибровочных графиков. При этом график зависимости аналитического сигнала от количества или концентрации определяемого вещества может быть аппроксимирован подходящей нелинейной функцией с использованием метода наименьших квадратов, что выполнимо при наличии соответствующего валидированного программного обеспечения.
ПРАВИЛЬНОСТЬ
Правильность методики характеризуется отклонением среднего результата определений, выполненных с ее использованием, от значения, принимаемого за истинное.
Валидируемая методика признается правильной, если значения, принимаемые за истинные, лежат внутри доверительных интервалов соответствующих средних результатов анализов, полученных экспериментально по данной методике.
Для оценки правильности методик количественного определения применимы следующие подходы:
- а) анализ с использованием валидируемой методики стандартных образцов или модельных смесей с известным содержанием (концентрацией) определяемого вещества;
- б) сравнение результатов, полученных с использованием валидируемой методики и образцовой методики, правильность которой ранее установлена;
- в) рассмотрение результатов изучения линейности валидируемой методики: если свободный член в уравнении, приведенном в разделе 5, статистически достоверно не отличается от нуля, то использование такой методики дает результаты, свободные от систематической ошибки.
Для подходов «а» и «б» возможно представление полученных данных в виде уравнения линейной зависимости (регрессии) между экспериментально найденными и истинными величинами. Для этого уравнения проверяются гипотезы о равенстве единице тангенса угла наклона b и о равенстве нулю свободного члена a. Как правило, если эти гипотезы признаются верными при степени надежности, равной 0,05, то использование валидируемой методики дает правильные, т. е. свободные от систематической ошибки, результаты.
ПРЕЦИЗИОННОСТЬ
Прецизионность методики характеризуется рассеянием результатов, получаемых с ее использованием, относительно величины среднего результата. Мерой такого рассеяния является величина стандартного отклонения результата отдельного определения, полученная для выборки достаточно большого объема.
Прецизионность оценивается для любой методики количественного определения по результатам не менее трех определений для каждого из трех уровней определяемых величин (нижнего, среднего и верхнего), лежащих в пределах аналитической области методики. Повторяемость также может оцениваться для любой методики количественного определения по результатам не менее шести определений для образцов с содержанием определяемого вещества, близким к номинальному. Во многих случаях оценка прецизионности может быть проведена по результатам обработки экспериментальных данных методом наименьших квадратов, как указано в ОФС «Статистическая обработка результатов химического эксперимента».
Прецизионность должна исследоваться на однородных образцах и может оцениваться в трех вариантах:
- – как повторяемость (сходимость);
- – как внутрилабораторная (промежуточная) прецизионность;
- – как межлабораторная прецизионность (воспроизводимость).
Результаты оценки методики анализа по каждому из вариантов прецизионности обычно характеризуются соответствующим значением величины стандартного отклонения результата отдельного определения.
Обычно при разработке оригинальной методики определяется повторяемость (сходимость) результатов, получаемых с ее использованием. При необходимости включения разработанной методики в нормативную документацию дополнительно определяется ее внутрилабораторная (промежуточная) прецизионность. Межлабораторная прецизионность (воспроизводимость) методики оценивается при предполагаемом ее включении в проект общей фармакопейной статьи, фармакопейной статьи или в нормативную документацию на фармакопейные стандартные образцы.
7.1. Повторяемость (сходимость)
Повторяемость аналитической методики оценивают по независимым результатам, полученным в одинаковых регламентированных условиях в одной лаборатории (один и тот же исполнитель, одно и то же оборудование, один и тот же набор реактивов) в пределах короткого промежутка времени.
7.2. Внутрилабораторная (промежуточная) прецизионность
Внутрилабораторная (промежуточная) прецизионность валидируемой методики оценивается в условиях работы одной лаборатории (разные дни, разные исполнители, разное оборудование и т. д.).
7.3. Межлабораторная прецизионность (воспроизводимость)
Межлабораторная прецизионность (воспроизводимость) валидируемой методики оценивается при проведении испытаний в разных лабораториях.
УСТОЙЧИВОСТЬ
Устойчивость валидируемой методики – это способность сохранять найденные для нее в оптимальных (номинальных) условиях характеристики, приведенные в таблице, при вероятных небольших отклонениях от этих условий проведения анализа.
Устойчивость методики не следует определять по отношению к легко контролируемым условиям проведения анализа. Это резко сокращает необходимость в специальном изучении устойчивости.
Устойчивость должна изучаться только в тех случаях, когда валидируемая методика основана на использовании особо чувствительных к внешним условиям методов анализа, таких как различные виды хроматографии и функционального анализа. При необходимости оценка устойчивости методики проводится на стадии ее разработки. Если вероятна невысокая устойчивость методики, проверка ее пригодности осуществляется в обязательном порядке непосредственно в процессе практического использования.
Проверка пригодности аналитической системы
Проверка пригодности аналитической системы – это проверка выполнения основных требований, предъявляемых к ней. Система, пригодность которой проверяется, представляет собой совокупность конкретных приборов, реактивов, стандартов и анализируемых образцов. Требования к такой системе обычно конкретизированы в общей фармакопейной статье на соответствующий аналитический метод. Таким образом, проверка пригодности аналитической системы становится процедурой, включаемой в валидируемую методику.
Представление результатов валидации
Протокол валидации аналитической методики должен содержать:
- – ее полное описание, достаточное для воспроизведения и отражающее все условия, необходимые для выполнения анализа;
- – оцениваемые характеристики;
- – все первичные результаты, которые вошли в статистическую обработку данных;
- – результаты статистической обработки данных, полученных экспериментально при разработке или проверке валидируемой методики;
- – иллюстративные материалы, такие как копии хроматограмм, полученных методами высокоэффективной жидкостной хроматографии или газовой хроматографии; электрофореграмм, электронных и инфракрасных спектров; фотографии или рисунки хроматограмм, полученных методами тонкослойной или бумажной хроматографии; рисунки кривых титрования, калибровочные графики;
- – заключение о пригодности валидируемой методики для включения в нормативный документ.
Материалы валидации отдельных аналитических методик целесообразно оформлять в виде объединенного отчета о валидации.