An evolutionary or phylogenetic tree both have the same names. It is a branching diagram or tree that represents the relationships that have developed over time between different biological species or other entities based on the similarities and differences in their physical or genetic traits. One phylogenetic tree, which shows a common ancestor for all life on Earth, is present.
Human Phylogenetic Tree
Every species or person (in this example) has a common ancestor, as shown in the diagram, and that person is your grandparent. Then it separates into your parent’s and your aunt’s branches (sibling of your parent). Because you were born to different parents yet have a similar ancestor to your grandparent, you, your sibling, and your cousins have a special history.

History
Ancient beliefs of a ladder-like evolution from lower to higher life forms gave rise to the concept of a “tree of life” (such as in the Great Chain of Being). A “paleontological chart” outlining the geological relationships between plants and animals can be found in Edward Hitchcock’s book Elementary Geology as one of the earliest examples of “branching” phylogenetic trees (first edition: 1840). In his ground-breaking book The Origin of Species, Charles Darwin (1859) also created one of the first pictures and played a significant role in popularising the idea of an evolutionary “tree.” The concept that speciation occurs through the adaptive and semi-random splitting of lineages is successfully communicated by tree diagrams, which are still used by evolutionary biologists to represent evolution more than a century after they were first used. The taxonomy of species has evolved to become more dynamic and less static.
Parts of a Phylogenetic Tree
A phylogenetic tree consists of the following components:
- Every branch denotes a lineage (single line of descent).
- Each node on a branch (also known as a branch point) reflects the split in two or more evolutionary lineages from a common ancestor.
- A taxon (plural: taxa), which might be a species or a group at any hierarchical level, is represented by each leaf, also known as a terminal node.
- Sister taxa are groups of related taxa that diverge from a single node. They stand for species that have a more recent common ancestor than other groups. Sister taxa have the closest relationships among their members.
- Taxa close to the root are called basal taxa. They are examples of species or groups that, early in the course of their evolutionary histories, diverge from the other members of the group.
- The most recent common ancestor of all taxa is shown as the tree’s root. Some phylogenetic trees do not have roots.

Parts of a phylogenetic tree
Approaches to make Phylogenetic Tree
The phylogenetic tree is created using one of two different approaches:
Character-based Approach
This method is also known as the discrete method because it is based solely on the sequence characters. Aligned characters are used in the character-based technique to build the phylogenetic tree.
During the tree inference, these aligned characters either include DNA or protein sequences. Maximum parsimony and Character based approaches are the two most prevalent.
Distance-based Approach
This approach is based on how dissimilar or how far apart the two aligned sequences are from one another. The pairwise distances from the sequence data are then utilized to create a matrix, which is subsequently used to generate the phylogenetic tree in this method.
Steps for Phylogenetic Analysis
Any phylogenetic study starts with the following fundamental steps:
Step 1: Setup and alignment of a dataset
- Finding an interesting protein or DNA sequence is the initial stage, followed by compiling a dataset of related sequences.
- Using NCBI BLAST or other comparable search engines, DNA sequences of interest can be located.
- Multiple sequence alignment is produced after the selection and recovery of sequences.
- To find homology regions, a set of sequences must be arranged in a matrix.
- ClustalW, MSA, MAFFT, and T-Coffee are just a few of the websites and software tools available for doing multiple sequencing on a given set of molecular data.
Step 2: Create (estimate) phylogenetic trees
- From sequences using stochastic models and computational techniques.
- Statistical techniques are used to ascertain the tree topology and calculate the branch lengths that most accurately depict the phylogenetic relationships of the matched sequences in a dataset in order to construct phylogenetic trees.
- The most often used computational techniques are those that use distance matrices and discrete data, including maximum likelihood and parsimony.
- Many software programs, including Paup, PAML, and PHYLIP, use these most common techniques.
Step 3: Test and evaluate the estimated trees statistically.
- One or more ideal trees are produced via tree estimation techniques.
- A number of statistical tests are run on this set of potential trees to see which is the best option and whether the suggested phylogeny makes sense.
- Jackknife Resampling techniques, as well as analytical techniques like parsimony, distance, and likelihood, are frequently used to evaluate trees.
Types of Phylogenetic Tree
Distinct phylogenetic trees are divided into varied groups based on their different traits, such as whether they are rooted, non-rooted, bifurcating, or multifurcating.
- Rooted tree: A phylogenetic tree with a common ancestor on each node is referred to as a rooted tree. As a result, the categorization comes to a stop at one point, typically at the node that serves as the common ancestor of all the tree branches.
- Unrooted tree: The non-rooted tree does not share a common ancestor with the rooted tree. The common ancestor or the tree node is always left out while creating the unrooted phylogenetic tree from the rooted tree.
- Bifurcating tree: Phylogenetic trees that only have two branches or leaves are referred to as bifurcating trees. Additionally, it can be divided into rooted and unrooted bifurcating trees.
- Multifurcating tree: Multiple branches can be found on a single node in a multifurcating tree, as the name suggests. Both a rooted multifurcating tree and an unrooted multifurcating tree are categories for it once more.
It was once thought that multicellular eukaryotic beings descended from ape-like prokaryotes. The evolution process is governed by several forces. One of them is genetic makeup.
Based on morphological, genotypic, and phylogenetic variances and similarities across species, a phylogenetic tree is created.
This means that to create an accurate phylogenetic tree, both observable alterations and changes in DNA sequences are taken into consideration.

Special Types of the Phylogenetic Trees
- Dendrogram-A phylogenetic tree’s diagrammatic representation is also known as a dendrogram because a dendrogram is a broad term for any tree, phylogenetic or not.
- Cladogram-A cladogram solely depicts a branching pattern; as a result, its interior nodes do not represent ancestors and its branch lengths do not correspond to time or the relative degree of character change.
- Chronogram-A Chronogram is a particular kind of Phylogenetic tree that uses the length of its branches to represent time.
- Phylogram-A phylogenetic tree with branch lengths according to character change is called a phylogram. A phylogenetic tree called a chronogram explicitly displays time by the lengths of its branches.
- Dahlgrenogram-A Dahlgrenogram is a diagram that shows a phylogenetic tree in cross-section.
Importance
The most important data from the disciplines of anatomy, paleontology, molecular genetics, and embryology, may be derived using this essential method. The evolutionary tree also has the following significance:
- To illustrate the relationships between organisms thought to share some evolutionary origin.
- Researching the shared ancestors of extinct and surviving species.
- Employed to research the evolutionary past.
- Employed in the hunt for new species.
- The evolutionary histories of pathogenic bacteria can be tracked with the use of the phylogenetic tree.
- Research the global dispersal of the species
- It is used to determine the most recent shared ancestors and how closely related different species are to one another.
- To connect the important turning points in the development of life to the tree of life.
Applications
- The goal of the phylogenetic tree is to establish an evolutionary connection between distinct creatures. By doing this, we can learn more about the evolutionary processes and sources of various creatures.
- It is useful to investigate evolution-related occurrences and classify species according to how their structures and functions have diverged.
- Additionally, it is useful to organize organisms and species according to their DNA sequences and morphological similarities and differences.
- Studying the impacts of evolution and the traits of various organisms is also helpful.
Limitations
- This evolutionary tree of craniates, which resembles a progressing ladder, evolved from an organism without a spinal column.
- Therefore, depending only on the traits they share, various groupings of organisms, objects, or units are situated at the tips of each branch.
- A phylogenetic tree illustrates the theories regarding the evolution and development of life.
- They are only as accurate as the facts that they are based on and are supported by.
- The information is derived from research and studies, which may contain some bias.
- As a result, phylogenetic trees constructed using data from research and studies may always be erroneous, biased, or subject to manipulation.
Cladogram
A phylogeny, or hypothetical link between groups of creatures, is depicted in a diagram known as a cladogram. A phylogenetic systematics researcher will use a cladogram to depict the groups of organisms being compared, their relationships, and their most recent shared ancestors. A cladogram might be quite complicated and compare every known form of life, or it can be extremely simple and compare just two or three groups of creatures.
Difference between a Cladogram and a Phylogenetic Tree
Phylogenetic trees and cladograms are frequently used interchangeably; however, they differ in some ways.
Cladograms demonstrate the evolutionary relationships between various organisms without demonstrating the course of evolution. Through the evolutionary changes that have taken place between an ancestor and its descendant species or set of species, phylogenetic trees demonstrate how various organisms are connected.
FAQs on Phylogenetic Trees
Question 1: Define a phylogenetic tree.
Answer:
An evolutionary or phylogenetic tree both have the same names. It is a branching diagram or tree that represents the relationships that have developed over time between different biological species or other entities based on the similarities and differences in their physical or genetic traits
Question 2: Write the difference between a cladogram and a phylogenetic tree.
Answer:
Cladograms demonstrate the evolutionary relationships between various organisms without demonstrating the course of evolution. Through the evolutionary changes that have taken place between an ancestor and its descendant species or set of species, phylogenetic trees demonstrate how various organisms are connected.
Question 3: Write about the spindle diagram.
Answer:
A spindle diagram, with the breadth of the spindles denoting different families, illustrates the evolution of vertebrates at the class level. The usage of spindle diagrams in evolutionary taxonomy is common. After its popularisation by the American paleontologist Alfred Romer, a spindle diagram, also known as a bubble diagram, is frequently referred to as a Rome program. To reflect changes in taxon abundance across time, it plots taxonomic diversity (horizontal breadth) versus geological time (vertical axis).
Question 4: What is a cladogram?
Answer:
A cladogram solely depicts a branching pattern; as a result, its interior nodes do not represent ancestors and its branch lengths do not correspond to time or the relative degree of character change.
Question 5: Write the importance of a phylogenetic tree.
Answer:
- To illustrate the relationships between organisms thought to share some evolutionary origin.
- Researching the shared ancestors of extinct and surviving species.
- Employed to research the evolutionary past.
- Employed in the hunt for new species.
- The evolutionary histories of pathogenic bacteria can be tracked with the use of the phylogenetic tree.
Last Updated :
12 Oct, 2022
Like Article
Save Article
Филогенетическое дерево (эволюционное дерево, дерево жизни) — дерево, отражающее эволюционные взаимосвязи между различными видами или другими сущностями, имеющими общего предка.
Вершины филогенетического дерева делятся на три класса: листья, узлы и (максимум один) корень. Листья — это конечные вершины, то есть те, в которые входят ровно по одному ребру; каждый лист отображает некоторый вид живых организмов (или иной объект, подверженный эволюции, например, домен белка). Каждый узел представляет эволюционное событие: разделение предкового вида на два или более, которые в дальнейшем эволюционировали независимо. Корень представляет общего предка всех рассматриваемых объектов. Ребра филогенетического дерева принято называть «ветвями».
Идея «дерева» появилась в ранних взглядах на жизнь, как на процесс развития от простых форм к сложным. Современные эволюционные биологи продолжают использовать деревья для иллюстрации эволюции, так как оно наглядно показывает развитие и происхождение видов.
Типы филогенетических деревьев
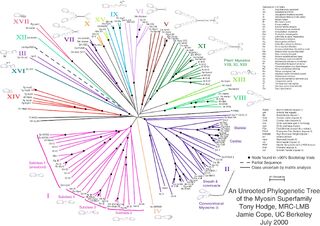
Рис. 1: Теоретически укоренённое дерево для генов рРНК Рис. 2: Неукоренённое дерево для семейства супергена миозина[1]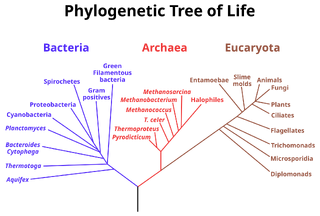
Укоренённое дерево — дерево, содержащее выделенную вершину — корень. Корневое дерево можно считать ориентированным графом, поскольку на нем имеется естественная ориентация — от корня к листьям. Каждый узел корневого дерева отвечает последнему общему предку нижележащих листьев дерева. Рисунок 1 представляет корневое филогенетическое дерево, окрашенное в соответствии с трёхдоменной системой живых организмов. [2].
Неукоренённое дерево не содержит корня и отражает связь листьев без предполагаемого положения общего предка. Необходимость рассматривать некорневые деревья возникает из-за того, что часто связи между узлами восстановить легче, чем направление эволюции. Рисунок 2 иллюстрирует некорневое филогетическое дерево. [3]. Наиболее достоверным методом для превращения неукорененного дерева в укорененное (для этого надо либо объявить корнем один из узлов, либо разбить одну из ветвей на две, выходящие из корня) является использование достоверной «внешней группы» видов — достаточно близких к интересующему нас набору видов для достоверного восстановления топологии дерева для объединенного множества видов, но в то же время заведомо являющихся отдельной группой. Иногда положение корня можно угадать, исходя из каких-либо дополнительных знаний о природе изучаемых объектов (видов, белков, etc.)
Укоренённое и неукоренённое филогетическое дерево может быть бифуркационным или небифуркационным, а также маркированным или немаркированным.
В бифуркационном дереве к каждому узлу подходят ровно три ветви (в случае корневого дерева — одна входящая ветвь и две исходящие). Таким образом бифуркационное дерево предполагает, что все эволюционные события состояли в происхождении от предкового объекта ровно двух потомков. К узлу небифуркационного дерева могут подходить четыре и более ветви.
Маркированное дерево содержит названия листьев, тогда как немаркированное просто отражает топологию.
Дендрограмма — общий термин, обозначающий схематическое представление филогенетического дерева.
Кладограмма — филогенетическое дерево, не содержащее информации о длинах ветвей.
Филограмма (или фенограмма) — филогенетическое дерево, содержащее информацию о длинах ветвей; эти длины представляют изменение некой характеристики.
Хронограмма — филограмма, длины ветвей в которой представляют эволюционное время.
Построение филогенетических деревьев
Филогенетические деревья из неограниченного числа входных последовательностей составляются используя вычислительные филогенетические методы. Методы матричного расстояния, такие как методы ближайшего вхождения, которые требуют множественные выравнивания цепочек для вычисления генетического расстояния, просты в применении; методы выравнивания множества цепочек, наподобие используемых в программе ClustalW выполняют как выравнивание цепочки, так и филогенетических деревьев. Другие методы максимально экономичны и используют приближённые технические приёмы такие, как максимальная вероятность; приближение Байеса также применимо к филогенетике, но оно спорно.[4] Нахождение оптимального дерева, используя многие из этих технических способов НП-полно[5] или НП-трудно[4], поэтому эвристический поиск и методы оптимизации используются в сочетании с функциями обсчёта дерева для нахождения хорошо подходящего дерева, удовлетворяющего входным данным.
Методы построения дерева могут быть оценены по нескольких основным критериям:[6]
- эффективность (насколько долго вычисление ответа, сколько памяти для этого потребуется?)
- производительность (есть ли польза от полученных данных или информация бесполезна?)
- постоянство (будут ли повторные ответы такими же, если каждый раз даются разные данные для той же проблемной модели?)
- устойчивость к ошибкам (справляется ли с нарушениями в предпосылках расссматриваемой модели?)
- выдача предупреждений (будет ли предупреждать нас, когда неправильно используется, т.е. предпосылки неверные?)
Также методы построения дерева могут быть предложены вниманию математиков. Деревья могут быть построены, используя Т-теорию.[7]
Ограниченность филогенетических деревьев
Хотя филогенетические деревья, построенные на основе генных цепочек или данных генома в различных видах особей могут дать представление об эволюции, у них есть серьёзные ограничения. Филогенетические деревья не обязательно (и вероятно никогда) не дают фактического представления об эволюционной истории. Данные, на которых они основываются, являются шумом; горизонтальная передача гена[8], гибридизация между видами, не являющимися близкородственными, конвергентная эволюция и сохранение цепочек — всё это может быть основой для анализа. Для избежания этих ограничений в программе PhyloCode есть один метод анализа, не предполагающий использование древовидной структуры.
Кроме того, существует проблема в анализе, основанном на единственном отличительном признаке, например, единственном гене или протеине или только на морфологическом анализе, потому что такие деревья, построенные на основе другого независимого источника данных, часто отличаются от первого, и поэтому много внимания надо уделять выведению филогенетических взаимосвязей между видами.
Это более всего справедливо для генетического материала, который является предметом горизонтальной передачи генов и их рекомбинации, при которой различные блоки гаплотипов могут быть с разной историей.
В основном, вывод дерева филогенетического анализа — это оценка филогении особенностей (то есть дерево гена), а не филогении таксона (то есть дерева видов), из которого были отобраны эти отличительные характерные особенности, хотя в идеале оба должны быть весьма близкими.
Когда вымершие виды включены в дерево, они являются конечными точками, поскольку маловероятно, чтобы они были прямыми предками любых существующих видов. Следует скептически относиться к включению в дерево вымерших видов, информация о которых полностью или частично основана на данных цепочки ДНК, на основе того факта, что небольшая полезная «древняя ДНК» сохраняется дольше 100 000 лет, и, за исключением необычных случаев, цепочка ДНК не является достаточно длинной для использования в филогенетических анализах, даже если она взята из материала давностью до 1 млн. лет.
В некоторых организмах эндосимбионты могут иметь генетическую историю, независимую от носителя.
Филогенетические сети используются, когда бифуркация деревьев не уместна, из-за этих сложностей охват эволюционной истории выбранных организмов имеет более сетчатый узор.
См. также
- Эволюция животных
- Дендрограмма
- Кладограмма
- Эволюционное учение
- Филогенетика
- Таксономия
- Генеалогическое дерево
Примечания
- ↑ Hodge, T. & M. J. T. V. Cope. 2000. A Myosin Family Tree. Journal of Cell Science 113: 3353-3354. See also the Myosin external link below.
- ↑ Woese, C. R. 1998. The Universal Ancestor. en:Proceedings of the National Academy of Sciences 95: 6854-6859.
- ↑ Maher, B. A. 2002. Uprooting the Tree of Life. The Scientist 16: 18 (Sep. 16, 2002); subscription only
- ↑ 4,0 4,1 Felsenstein J. (2004). Inferring Phylogenies Sinauer Associates: Sunderland, MA.
- ↑ Так говорят о классе комбинаторных задач с нелинейной полиномиальной оценкой числа итераций
- ↑ Penny, D., Hendy, M. D. & M. A. Steel. 1992. Progress with methods for constructing evolutionary trees. Trends in Ecology and Evolution 7: 73-79.
- ↑ A. Dress, K. T. Huber, and V. Moulton. 2001. Metric Spaces in Pure and Applied Mathematics. Documenta Mathematica LSU 2001: 121—139
- ↑ Woese, C. R. 2002. On the evolution of cells. Proceedings of the National Academy of Sciences 99: 8742-8747.
Изображения в интернете
- Phylogenetic Trees Based on 16s rDNA
- A 3D View
- Human Y-Chromosome 2002 Phylogenetic Tree
- In 2003, the Science journal dedicated a special issue to the tree of life, including an online version of a tree of life.
Общее описание
- PhyloCode
- A Multiple Alignment of 139 Myosin Sequences and a Phylogenetic Tree
- Tree of Life Web Project
- http://www.aisee.com/graph_of_the_month/jura.htm — The most detailed and comprehensive family tree of dinosaurs yet available
- http://www.omne-vivum.com tree of life with lots of pictures
Эта страница использует содержимое раздела Википедии на русском языке. Оригинальная статья находится по адресу: Филогенетическое дерево. Список первоначальных авторов статьи можно посмотреть в истории правок. Эта статья так же, как и статья, размещённая в Википедии, доступна на условиях CC-BY-SA .
1. Построение филогенетических деревьев
2. Особенности молекулярной эволюции
1. Скорость эволюции любого белка,
выраженная через число аминокислотных
замен на сайт в год, приблизительно
постоянна и одинакова в разных
филогенетических линиях, если только
функция и третичная структура этого белка
остаются в основном неизменными.
3. Что такое филогенетические деревья?
0.02
Gallus
Rattus
Mus
Дерево — это граф, в
котором два соседних узла
соединены только одним
ребром.
Bos
Homo
Xenopus
4. Кладограммы и филограммы
Кладограммы отражают только порядок
ветвления, филограммы — ещё и длину
ветвей
5. Сколько здесь разных кладограмм?
d
a
d
b
b
a
e
c
e
c
c
e
e
a
a
b
b
d
d
c
6. Выбор последовательностей
• Последовательности должны быть
гомологичны! Программа
выровняет любые
последовательности => нужно
проверить с помощью Blast
• Затем нужно выровнять
последовательности, и по
получившемуся выравниванию,
определить, какие
последовательности включить в
анализ
7. «Эффект тыквенного пирога»
Рецепт тыквенного
пирога на
филогенетическом
дереве креветок.
8. Выбор последовательностей
9. Особенности молекулярной эволюции
2. Функционально менее важные молекулы
или их части эволюционируют (накапливая
эволюционные замены) быстрее, чем более
важные
3. Мутационные замены, приводящие к
меньшим нарушениям структуры и
функции молекулы (консервативные
замены), в ходе эволюции происходят чаще
тех, которые вызывают существенное
нарушение структуры и функции этой
молекулы
10. Различия между деревом генов и деревом видов
Проблема: ортологи и паралоги
11. Молекулярная конвергенция
12. Филогенетические маркёры
Свойства:
• Гены, которые представлены одной
копией в геноме лучше, чем те, у которых
множество копий.
• Длина гена не должна варьировать у
разных организмов
• Скорость изменения гена должна
соответствовать скорости эволюции
таксонов заданного уровня
• Должны легко подбираться
специфические праймеры
13.
14. Филогенетические маркёры
• Рибосомальные гены
• Митохондриальные гены
(COI/II, 12s RNA, cyt b)
• Хлоропластные гены
• Гены домашнего
хозяйства и некоторые
другие ядерные
15. Выбор модели замен
Результаты вычисления эволюционных дистанций будут
отличаться в зависимости от выбранной модели замен
16. Выбор модели замен
AIC — Akaike’s Information Criterion. Быстрее
BIC — Bayesian information criteria. Не «любит» более сложные
модели
DT — decision theory
LRT — тест соотношения вероятностей. «Любит» более
сложные модели.
17.
18. Методы реконструкции филогении
Дистанционные
Максимальной
экономии
Максимальной
вероятности
Используют только
попарные дистанции
Используют только
символьные данные
Используют все данные
Минимизация
дистанции между
ближайшими соседями
Минимизация общей
длины дерева
(минимизация числа
мутаций
Максимизация
вероятности заданного
дерева с учётом заданных
параметров
Очень быстрые
Медленные
Очень медленные
Ищут локальный
оптимум вместо
глобального
Неверны при
быстрой скорости
эволюции
Сильно зависят от
правильности выбранной
модели
Хороши для чернового
или предварительного
дерева или выбора
между множеством
деревьев
Лучший выбор для
подходящей
выборки(<30
последовательностей,
без гомоплазий)
Хороши для очень
маленьких наборов
данных и для оценки
топологий, построенных
другими методами
19. Дистанционные методы Neghbor-joining
Начинаем с пары ветвей, которые
меньше всего отличаются между
собой
20. Дистанционные методы Neghbor-joining
21. Дистанционные методы Neghbor-joining
22. Зачем нужна аутгруппа
Молекулярнофилогенетические
методы используют
информацию о
последовательностях
внешней группы
(контроля), дистанция
от которой для всех
остальных
последовательностей
заведомо выше, чем от
других.
Таким образом дерево
«укореняется», а также
внутри дерева убирается
«шум»
23. Дистанционные методы Neghbor-joining
•Не учитываются обратные и параллельные замены
=> Мы считаем не настоящую дистанцию (расстояние), а
редакционное расстояние.
•Вычислительно более быстрые.
•В большинстве случаев оценивают только топологию дерева,
не воспроизводя исходную последовательность.
•Если у нас будет бесконечная последовательность, то мы с
вероятностью 100% получим истинное дерево.
24. Методы максимальной экономии
•Минимизация числа замен символов
•Всегда реконструируют предковые последовательности
•Лучше работает на
небольших наборах
последовательностей
во многих случаях
на больших объёмах
данных работает хуже.
25. Методы максимальной экономии
(2n 3)!
N =
R 2 n 2 (n 2)!
Число внешних
узлов (таксонов)
2
3
4
5
10
20
Число возможных
деревьев
1
3
15
105
34459425
8200794532637891559375
26.
27. Методы максимальной вероятности
•Так же, как и в случае с методами максимальной экономии,
генерирует все возможные топологии деревьев
•Предположение особой модели эволюции
•В отличие от метода максимальной экономии может
предполагать разную скорость эволюции и скорость замен в
разных ветвях дерева
•Поиск дерева с максимальной вероятностью существования,
соответствующего данным
•Чем больше последовательность, тем вероятнее найти
истинное дерево
•Самые медленные
28. Методы максимальной вероятности
• В позиции j для каждого внутреннего узла
допустимы все четыре нуклеотида, значит
всего 4*4=16 возможных деревьев.
• Каждое из деревьев это произведение
вероятности возникновения какого-либо
основания в корне дерева и вероятность его
замены на тот, который в следующем узле.
Т.е. частота нуклеотида умноженная на
вероятность его мутации, если грубо.
A = 0.25 or средняя частота A в
последовательности зависит от модели) A->C
трансверсия = 10-6 and A->G транзиции = 2×10-6
Вероятность T1 = 0.25 x 2×10-6 x 10-6 = 5×10-13
• Вероятность всего дерева равна
произведению вероятностей деревьев для
каждой позиции в выравнивании
29. Оценка поддержки дерева
•Bootstrap
001122234556667
rat
GGAAGGGGCTTTTTA
human
GGTTGGGGCTTTTTA
turtle
GGTTGGGCCCCTTTA
fruitfly CCTTCCCGCCCTTTT
oak
AATTCCCGCTTCCCT
duckweed AATTCCCCCTTCCCC
0123456789
rat
GAGGCTTATC
human
GTGGCTTATC
turtle
GTGCCCTATG
fruitfly CTCGCCTTTG
oak
ATCGCTCTTG
duckweed ATCCCTCCGG
445556777888899
rat
CCTTTTAAATTTTCC
human
CCTTTTAAATTTTCC
turtle
CCCCCTAAATTTTGG
fruitfly CCCCCTTTTTTTTGG
oak
CCTTTCTTTTTTTGG
duckweed CCTTTCCCCGGGGGG
rat
human
turtle
fruit fly
oak
duckweed
Повторить
перестановку
100 – 1000 раз
100
65
0
55
Inferred tree
rat
human
turtle
fruit fly
oak
duckweed
30. Оценка поддержки дерева
•Bayes inference

Добавил:
Вуз:
Предмет:
Файл:
Потапов В.В. Решение задач биоинформатики при помощи веб — и интернет-сервисов.pdf
Скачиваний:
80
Добавлен:
14.09.2020
Размер:
2.93 Mб
Скачать
Проанализируем последовательности первого кластера — сюда вошли последовательности таких вирусов, как Японский энцефалит (Japanese encephalitis virus), лихорадка Западного Нила, лихорадка Денге (West Nile virus), желтая лихорадка (Yellow fever virus) и другие. Интересно заметить, что все эти дальние родственники вызывают похожие заболевания у человека как и клещевой энцефалит, но распространены в более жарких странах и переносятся комарами. Это значит что человек заболевает после укуса комара, а укусы комаров как известно бывают гораздо чаще укусов клеща.
Филогения с древне-греческого дословно переводится phylon — «племя, раса» и genetikos — «имеющий отношение к рождению», в более широком смысле означает историческое развитие организмов.
В биологии филогенез рассматривает развитие биологического вида во времени.
Макромолекулярные данные, под которыми имеется в виду последовательности генетического материала ДНК и белков, накапливаются всё быстрыми темпами благодаря успехам молекулярной биологии. Для эволюционной биологии быстрое накопление данных последовательностей целых геномов имеет значительную ценность, потому что сама природа ДНК позволяет использовать его как «документ» эволюционной истории. Сравнения нуклеотидных или аминокислотных последовательностей у разных организмов могут сказать ученому много нового об эволюционных взаимоотношениях этих организмов, которые не могут быть обнаружены иначе, например, на основе морфологии, или внешней форме организмов, или их внутренней структуре. Поскольку геномы эволюционируют через постепенное накопление мутаций, количество отличий последовательности нуклеотидов между парой геномов разных организмов должно указать, как давно эти два генома отделились от общего предка. Два генома, которые разделились в недавнем прошлом, должны иметь меньшие отличий, чем два генома, чей общий предок более древний. Потому, сравнивая разные биологические последовательности друг с другом, возможно получить сведения об эволюционном взаимоотношения между ними. Это является главной задачей молекулярной филогенетики.
33
Молекулярная филогенетика пытается определить скорость и отличия изменений в ДНК и белках, чтобы восстановить эволюционную историю генов и организмов.
Задачей филогенетического анализа является установление, реконструкция эволюционной истории — родственных связей, отношений между формами жизни — и датирование эволюционных событий. В филогенетических исследованиях эволюционные отношения между формами жизни представляют в виде
филогенетических, или эволюционных, деревьев (phylogenetic, evolutionary trees).
Филогенетическое дерево состоит из внутренних и внешних ветвей (branches), узлов (nodes), и, если исследователем выбрана соответствующая опция, — корня (root), без корня (unroot). Порядок всех ветвей дерева называют его топологией (topology). Внутренние ветви соединяют внутренние узлы, внешние ветви ведут непосредственно к объектам исследования (также они называются внешние узлы, или листья дерева, leaves). Деревья с корнем отражают направление эволюции, порядок почкования, ответвления (branching) различных эволюционных линий.
Объектами филогенетического исследования могут быть гены или их участки, нуклеотидные или аминокислотные последовательности, организмы, популяции, индивидуумы, штаммы вирусов и т.д. Эти объекты исследования называют оперативными таксономическими единицами, OTU (Operational Taxonomic Units).
34
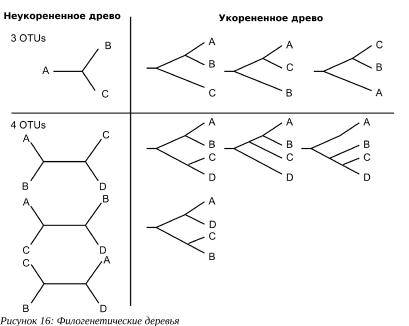
Деревья без корня показывают родственные отношения между анализируемыми последовательностями, но не направление эволюции.
После того как мы имеем представление о большой «семье» флавивирусов (род Flavivirus), не лишним будет построить филогенетическое древо, по топологии которого наглядно видны эволюционные расстояния между видами.
Порядок построения филогенетического древа:
1. Вернемся к выполненному запросу со скрининга по гомологии (GenBank), для этого удобно перейти по ссылке из письма о готовом задании, хранящемся у вас в электронной почте.
2.На странице с результатами выберите любые понравившиеся вам строки списка с неповторяющимися названиями вируса. Для того чтобы выбрать строку из списка, поставьте галочку, соответствующую этой строке.
35
3.Когда вы выберите все интересующие вас последовательности чтобы построить филогенетическое древо щелкните внизу на кнопку «Множественное выравнивание».
Так вы сможете воспользоваться сервисом который выравнивает заданный набор последовательностей, то есть расставляет гэпы так чтобы похожие участки в разных последовательностях находились одни под другим.
Для выполнения выравнивания нажмите на кнопку «рассчитать» с теми последовательностями во входной форме, которые туда были внесены когда вы перешли со страницы с результатами скрининга.;
4.После того как последовательности будут выровнены, щелкните внизу страницы с результатом выравнивания на ссылку «Перейти к филогенетическому анализу»
5.Через встроенную на сайте систему автоматических переходов (которые по английски называются pipelines) сформированный вами запрос перенаправляется на сервис Филогенетический анализ, здесь вы можете выбрать метод анализа и скомандовать «Рассчитать».
6.Результат построения филогенетического древа появляется практически мгновенно и вы сможете оценить эволюционные расстояния внутри выбранной вами группы вирусов и топологию построенного дерева.
5.2.4Конструирование трехмерной структуры вирусного белка NS3
Все задания выполненные до этого момента, были некоторым экскурсом для знакомства с исследуемым нами объектом, а теперь нам предстоит захватывающий этап работы, называемый Insight into, то есть взгляд внутрь. Давайте немного поразмышляем, над нашим объектом и устройством всего живого в целом. Из специализированных баз данных мы можем извлечь генетические последовательности практически для всех живых организмов живущих на Земле. Но что из себя представляет сама последовательность с точки зрения живой природы? Это — определенная матрица с которой синтезируется множество белков, выполняющих свои особенные исключительные функции в живом
36
организме. А благодаря белкам и существует живая материя. Как по настоящему выглядят белки, белки из которых состоит наше тело, или белки вирусной частицы, или любые другие белки? Для того чтобы узнать как выглядят белки в живой природе, существует несколько достаточно сложных экспериментальных методов — получение кристаллов белка и расшифровка их структуры с помощью рентгеноструктурного анализа и метод ядерно-магнитного резонанса, а так же компьютерное молекулярное моделирование.
Метод компьютерного молекулярного моделирования, в отличии от экспериментальных методов, позволяет получать лишь предсказания структуры белков, и эти предсказанные структуры могут отличаться от экспериментально наблюдаемых. По сути они представляют собой результаты эксперимента, который выполняется in silico, то есть на компьютере. Именно поэтому на сегодняшний день с помощью моделирования можно получить пространственную структуру белка достаточно быстро и дешево, хотя без дополнительной верификации такая структура будет отличаться от реального белка в растворе. В зависимости от требований к точности пространственной структуры выбираются разные методы либо долгие и дорогостоящие исследования с кристаллизацией белка, либо быстрые и малозатратные компьютерные эксперименты.
У нас есть последовательность вирусного белка NS3 по которой мы хотим воссоздать его третичную структуру, то есть увидеть как этот белок выглядит в природе.
Для этого воспользуемся известным методом моделирования по гомологии, этот метод также называется гомологичное моделирование, либо сравнительное моделирование.
Вкратце, суть метода можно описать так: существует база данных экспериментально определенных структур белков. Мы собираемся найти в этой базе структуру белка, который имеет гомологию по первичной последовательности с нашим заданным белком, и на основании найденной гомологии построить трехмерную модель нашего белка.
Несмотря на то что в базе данных PDB хранится огромное множество трехмерных моделей для самых разных белков, не всегда можно найти расшифрованную пространственную структуру непосредственно интересующего вас белка. Например, для исследуемого белка NS3 вируса клещевого энцефалита в PDB в настоящее время нет ни одной структуры. Однако если продолжить поиск в базе PDB можно найти
37

структуры этого же белка для близкородственных видов вируса того же рода и семейства. Если в базе белковых структур мы нашли хотя бы одну модель близкородственного белка, то можно приступать к моделированию по гомологии. Для выполнения задания по конструированию пространственной структуры вирусного белка NS3 воспользуемся сервисом «скрининг по гомологии (PDB)» на www.brishur.com
1.Откройте страницу сервиса Скрининг по гомологии (PDB)
2.По аналогии с предыдущими запросами заполните поля email и наименование задания Возьмите исследуемую последовательность белка NS3 и вставьте в поле «Последовательность»
3.Для уменьшения времени расчетов воспользуемся параметрами, заданными по умолчанию:
4.Метод скрининга для построения профиля Genebee
5.Количество последовательностей в полученном результате 100.
6.Cкомандуем «Рассчитать».
Рисунок 17: Bri-shur — скрининг по гомологии (PDB)
38

7.После выполнения задания в полученном вами письме перейдите по ссылке на страницу с результатом вашего запроса.
8.На странице со списком найденных гомологов выберите элемент списка с наибольшим значением Z-score
9.Щелкните внизу страницы на кнопку «Уточнить парное выравнивание»
10.Через систему пайплайнов вы окажетесь на сервисе множественное выравнивание.
11.Cкомандуем «Рассчитать».
12.Посмотрите на результат выравнивания и зрительно оцените процент сходства и совпадения исследуемой и найденной последовательностей, ориентируясь на количество условных символов * или +
13.Внизу страницы перейдите по ссылке «Перейти к моделированию по гомологии»
Рисунок 18: Bri-shur — задание параметров моделирования по гомологии в Nest
14.На странице «Nest (Гомологичное моделирование)» можно еще раз посмотреть выравнивание, щелкнув по кнопке «Предварительный просмотр выравнивания»
39
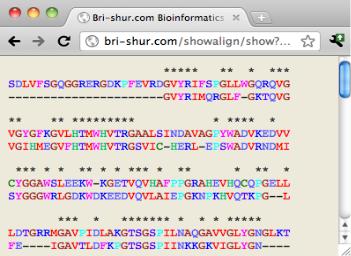
Рисунок 19: Bri-shur — пример выровненных последовательностей
15.Скомандуем «Рассчитать».
16.Через минуту обновите страницу и если задание выполнено щелкните на ссылку download и загрузите готовую трехмерную модель структуры белка в формате файла pdb на свой компьютер.
17.Далее визуализируйте модель с помощью программы Chimera, описанной выше.
40
Соседние файлы в предмете Биоинформатика
- #
- #
- #
- #
- #
16.09.202046.01 Mб11Павлов А.Н., Ермолаев Ю.М. Биоинформатика
- #
- #