Содержание
- 1 Краткая характеристика алканов
- 2 Галогенирование алканов
- 2.1 Стадии галогенирования
- 3 Региоселективность
- 3.1 Хлорирование и бромирование пропана
- 3.2 Галогенирование бутана
- 4 Реакция отщепления
- 5 Применение галогеналканов
Галогенирование – процесс присоединения к молекуле органического соединения атома галогена – это одно из химических взаимодействий, в которых участвуют углеводороды ряда алканов. Благодаря общности структуры молекул у всех членов гомологического ряда реакции с галогеном обладают единым характером. При этом галогенирование пропана и следующих за ним в гомологическом ряду соединений имеет особенности, связанные с наличием в молекулах вторичных, третичных и четвертичных атомов углерода.
Краткая характеристика алканов
К простейшим органическим соединениям относятся ациклические насыщенные, или предельные углеводороды (алканы). Их отличают следующие структурные особенности:
- неразветвленные (прямые) или разветвленные молекулы, не содержащие циклических образований;
- все химические связи в молекуле – одинарные.
Общая формула имеет вид ![]() . Углеродная цепь в молекулах алканов характеризуется полным насыщением водородными атомами, вследствие чего эти соединения проявляют слабую химическую активность. Они вступают в реакции замещения, окисления, разложения и изомеризации.
. Углеродная цепь в молекулах алканов характеризуется полным насыщением водородными атомами, вследствие чего эти соединения проявляют слабую химическую активность. Они вступают в реакции замещения, окисления, разложения и изомеризации.
Механизм замещения у алканов носит радикальный характер, так как разрыв слабополярной связи C-H протекает с образованием пары свободных радикалов – нейтральных частиц, имеющих по одному неспаренному электрону. Атом водорода может замещаться галогеном, нитрогруппой или сульфогруппой.
Галогенирование алканов
Реакция свободно-радикального замещения (обозначается символом SR – от англ. substitution radical reaction) водородного атома в молекуле алкана атомом галогена называется галогенированием. Ее инициирование требует воздействия ультрафиолетового облучения или повышенной температуры. Реакция носит цепной характер, так как в каждом ее акте образуются свободные радикалы, генерирующие следующий акт.
Замещение фтором – фторирование – чрезвычайно экзотермический процесс, сопровождающийся взрывом и разрушением молекулы алкана. Реакция с участием йода, наоборот, является эндотермической и обратимой – в ходе ее происходит восстановление продукта реакции йодоводородом, поэтому прямое йодирование неэффективно. Практический интерес представляют реакции алканов с хлором и бромом.

Стадии галогенирования
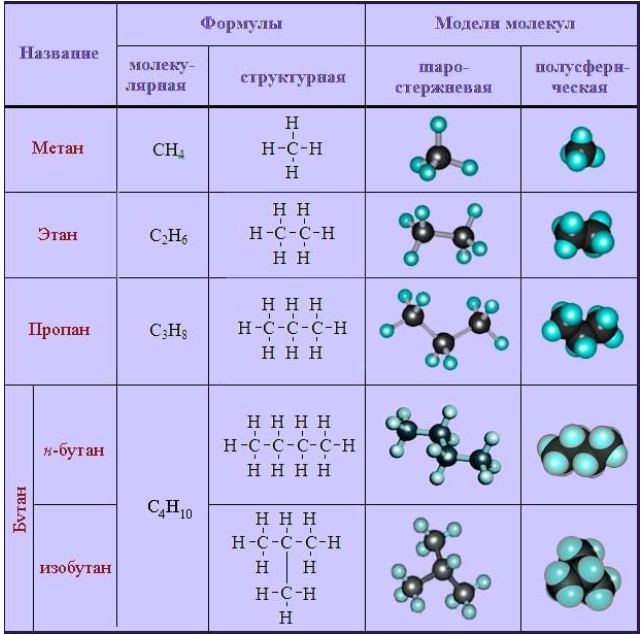
Удобнее всего рассмотреть, какой механизм имеет реакция хлорирования алканов, на примере метана. При облучении или нагреве реакционной смеси реакция инициируется, и далее цепная реакция протекает в несколько стадий.
- Зарождение цепи – распад молекулы хлора на активные радикалы:

- Развитие цепи. Взаимодействуя с молекулой метана
 , радикал
, радикал  отщепляет от нее атом водорода с образованием метил-радикала
отщепляет от нее атом водорода с образованием метил-радикала  , который, в свою очередь, расщепляет другую молекулу хлора. Эти элементарные акты повторяются многократно, образуя новые радикалы и развивая цепную реакцию:
, который, в свою очередь, расщепляет другую молекулу хлора. Эти элементарные акты повторяются многократно, образуя новые радикалы и развивая цепную реакцию:
- Обрыв цепи. Цепной процесс прекращается, когда радикалы реагируют между собой:


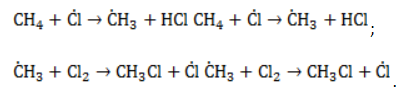

В общем виде уравнение галогенирования метана хлором записывается в форме:
![]()
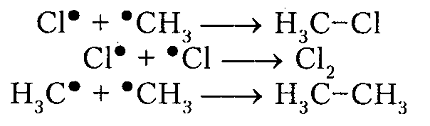
Процесс замещения не ограничивается образованием хлорметана, на молекулы которого также воздействуют радикалы ![]() . Реакция приводит к образованию смеси всех возможных продуктов хлорирования метана:
. Реакция приводит к образованию смеси всех возможных продуктов хлорирования метана:
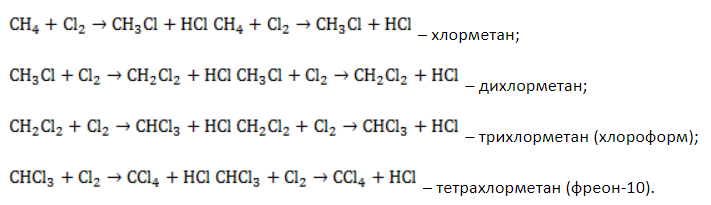
Выход продуктов зависит от мольного соотношения реагентов и условий реакции. Так, при эквимолярном количестве метана и хлора и температуре 440° C соотношение молярных долей продуктов в процентах составляет около 39:41:19:1.
Реакция бромирования отличается меньшим выделением энергии и протекает медленнее, так как его реакционная способность ниже, чем у хлора.
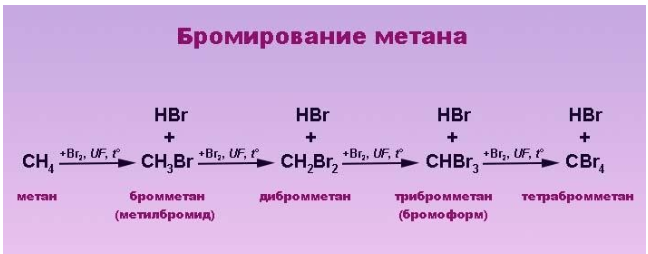
Региоселективность
Начиная с пропана ![]() , в составе молекул алканов появляются вторичные атомы углерода, а с бутана, имеющего два изомера (н-бутан
, в составе молекул алканов появляются вторичные атомы углерода, а с бутана, имеющего два изомера (н-бутан ![]() и изобутан
и изобутан ![]() ), – третичные, связанные соответственно с двумя и тремя другими углеродными атомами. Скорость галогенирования у разных атомов различна и возрастает в ряду «первичный → вторичный → третичный». Это явление носит название регионаправленности галогенирования, или региоселективности. Четвертичные атомы не участвуют в реакции радикального замещения.
), – третичные, связанные соответственно с двумя и тремя другими углеродными атомами. Скорость галогенирования у разных атомов различна и возрастает в ряду «первичный → вторичный → третичный». Это явление носит название регионаправленности галогенирования, или региоселективности. Четвертичные атомы не участвуют в реакции радикального замещения.
Селективность зависит от следующих факторов:
- Активность реагента. Чем активнее галоген, тем слабее проявляется избирательность при замещении водорода. Так, при взаимодействии алкана с хлором региоселективность значительно меньше, чем в реакциях с бромом, или не наблюдается.
- Температура. Нагревание ведет к снижению селективности.
Это интересно:
Общая формула циклоалканов
Изомеры гексана
Хлорирование и бромирование пропана
В реакции пропана с хлором при низкой температуре селективная направленность выражена слабо. Несмотря на то, что образующиеся в ходе реакции радикалы ![]() менее энергичны и, следовательно, более устойчивы, свободные атомы хлора чрезвычайно активны и воздействуют как на вторичные, так и на первичные атомы, особенно при высокой температуре.
менее энергичны и, следовательно, более устойчивы, свободные атомы хлора чрезвычайно активны и воздействуют как на вторичные, так и на первичные атомы, особенно при высокой температуре.
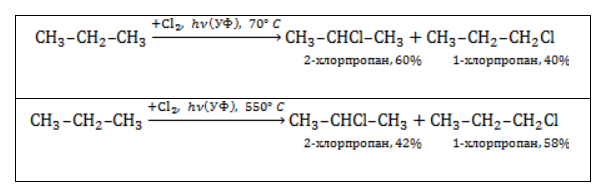
При нагревании радикалы хлора атакуют первичные атомы даже более активно, так как на разрыв их связи с водородом затрачивается меньше энергии.
При бромировании пропана региоселективность вследствие меньшей активности брома достигает высоких значений:

Бромирование протекает аналогично реакции с участием простейших алканов с преимущественным замещением водорода у вторичных атомов:
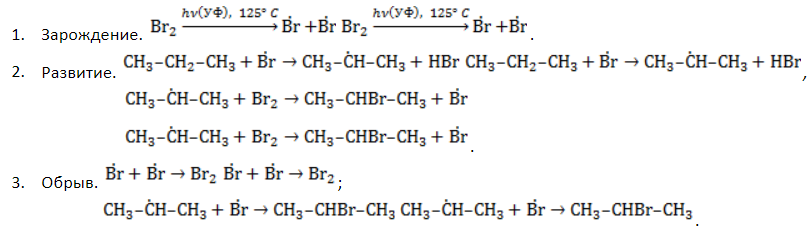
Продуктом этой реакции является 2-бромпропан.
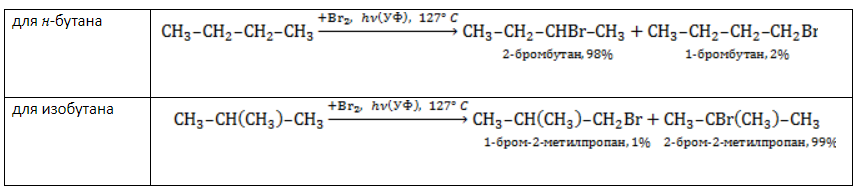
Галогенирование бутана
В реакциях бутана с хлором селективность не играет заметной роли. Даже при низкой температуре соотношение продуктов хлорирования может быть различным:

Бромирование бутана демонстрирует большую селективность:
Реакция отщепления
Активные двухвалентные металлы (как правило, магний или цинк) отщепляют от молекул дигалогеналканов атомы хлора или брома, если они замещают водород у двух соседних атомов углерода. Между последними образуется двойная связь. Продуктом такой реакции является алкен.
Пример реакции дегалогенирования алканов – отщепление цинком атомов хлора от 1,2-дихлорпропана с образованием пропилена (пропена) и хлорида цинка:
![]()
Применение галогеналканов
Хлорированные и бромированные алканы применяются в качестве промежуточных соединений в различных отраслях, таких как синтез высокомолекулярных соединений, производство лаков, красок и растворителей. Хлоралканы служат сырьем для фторалканов, которые нельзя получить прямым фторированием.
Токсичность галогеналканов тем меньше, чем активнее входящий в их состав галоген. Поэтому фторалканы наиболее безопасны. Фторсодержащие фреоны широко используются в качестве вспенивателей, хладагентов и пропеллентов.
Галогенирование алканов на примере пропана
Галогенирование – процесс присоединения к молекуле органического соединения атома галогена – это одно из химических взаимодействий, в которых участвуют углеводороды ряда алканов. Благодаря общности структуры молекул у всех членов гомологического ряда реакции с галогеном обладают единым характером. При этом галогенирование пропана и следующих за ним в гомологическом ряду соединений имеет особенности, связанные с наличием в молекулах вторичных, третичных и четвертичных атомов углерода.
Краткая характеристика алканов
К простейшим органическим соединениям относятся ациклические насыщенные, или предельные углеводороды (алканы). Их отличают следующие структурные особенности:
- неразветвленные (прямые) или разветвленные молекулы, не содержащие циклических образований;
- все химические связи в молекуле – одинарные.
Общая формула имеет вид 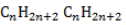 . Углеродная цепь в молекулах алканов характеризуется полным насыщением водородными атомами, вследствие чего эти соединения проявляют слабую химическую активность. Они вступают в реакции замещения, окисления, разложения и изомеризации.
. Углеродная цепь в молекулах алканов характеризуется полным насыщением водородными атомами, вследствие чего эти соединения проявляют слабую химическую активность. Они вступают в реакции замещения, окисления, разложения и изомеризации.
Механизм замещения у алканов носит радикальный характер, так как разрыв слабополярной связи C-H протекает с образованием пары свободных радикалов – нейтральных частиц, имеющих по одному неспаренному электрону. Атом водорода может замещаться галогеном, нитрогруппой или сульфогруппой.
Галогенирование алканов
Реакция свободно-радикального замещения (обозначается символом SR – от англ. substitution radical reaction) водородного атома в молекуле алкана атомом галогена называется галогенированием. Ее инициирование требует воздействия ультрафиолетового облучения или повышенной температуры. Реакция носит цепной характер, так как в каждом ее акте образуются свободные радикалы, генерирующие следующий акт.
Замещение фтором – фторирование – чрезвычайно экзотермический процесс, сопровождающийся взрывом и разрушением молекулы алкана. Реакция с участием йода, наоборот, является эндотермической и обратимой – в ходе ее происходит восстановление продукта реакции йодоводородом, поэтому прямое йодирование неэффективно. Практический интерес представляют реакции алканов с хлором и бромом.
Стадии галогенирования
Удобнее всего рассмотреть, какой механизм имеет реакция хлорирования алканов, на примере метана. При облучении или нагреве реакционной смеси реакция инициируется, и далее цепная реакция протекает в несколько стадий.
- Зарождение цепи – распад молекулы хлора на активные радикалы:

- Развитие цепи. Взаимодействуя с молекулой метана , радикал отщепляет от нее атом водорода с образованием метил-радикала , который, в свою очередь, расщепляет другую молекулу хлора. Эти элементарные акты повторяются многократно, образуя новые радикалы и развивая цепную реакцию:
- Обрыв цепи. Цепной процесс прекращается, когда радикалы реагируют между собой:
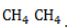 , радикал
, радикал 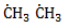 , который, в свою очередь, расщепляет другую молекулу хлора. Эти элементарные акты повторяются многократно, образуя новые радикалы и развивая цепную реакцию:
, который, в свою очередь, расщепляет другую молекулу хлора. Эти элементарные акты повторяются многократно, образуя новые радикалы и развивая цепную реакцию:В общем виде уравнение галогенирования метана хлором записывается в форме:
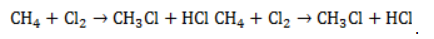
Процесс замещения не ограничивается образованием хлорметана, на молекулы которого также воздействуют радикалы 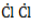 . Реакция приводит к образованию смеси всех возможных продуктов хлорирования метана:
. Реакция приводит к образованию смеси всех возможных продуктов хлорирования метана:
Выход продуктов зависит от мольного соотношения реагентов и условий реакции. Так, при эквимолярном количестве метана и хлора и температуре 440° C соотношение молярных долей продуктов в процентах составляет около 39:41:19:1.
Реакция бромирования отличается меньшим выделением энергии и протекает медленнее, так как его реакционная способность ниже, чем у хлора.
Региоселективность
Начиная с пропана 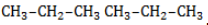 , в составе молекул алканов появляются вторичные атомы углерода, а с бутана, имеющего два изомера (н-бутан
, в составе молекул алканов появляются вторичные атомы углерода, а с бутана, имеющего два изомера (н-бутан 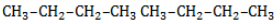 и изобутан
и изобутан 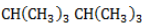 ), – третичные, связанные соответственно с двумя и тремя другими углеродными атомами. Скорость галогенирования у разных атомов различна и возрастает в ряду «первичный → вторичный → третичный». Это явление носит название регионаправленности галогенирования, или региоселективности. Четвертичные атомы не участвуют в реакции радикального замещения.
), – третичные, связанные соответственно с двумя и тремя другими углеродными атомами. Скорость галогенирования у разных атомов различна и возрастает в ряду «первичный → вторичный → третичный». Это явление носит название регионаправленности галогенирования, или региоселективности. Четвертичные атомы не участвуют в реакции радикального замещения.
Селективность зависит от следующих факторов:
- Активность реагента. Чем активнее галоген, тем слабее проявляется избирательность при замещении водорода. Так, при взаимодействии алкана с хлором региоселективность значительно меньше, чем в реакциях с бромом, или не наблюдается.
- Температура. Нагревание ведет к снижению селективности.
Хлорирование и бромирование пропана
В реакции пропана с хлором при низкой температуре селективная направленность выражена слабо. Несмотря на то, что образующиеся в ходе реакции радикалы 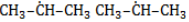 менее энергичны и, следовательно, более устойчивы, свободные атомы хлора чрезвычайно активны и воздействуют как на вторичные, так и на первичные атомы, особенно при высокой температуре.
менее энергичны и, следовательно, более устойчивы, свободные атомы хлора чрезвычайно активны и воздействуют как на вторичные, так и на первичные атомы, особенно при высокой температуре.
При нагревании радикалы хлора атакуют первичные атомы даже более активно, так как на разрыв их связи с водородом затрачивается меньше энергии.
При бромировании пропана региоселективность вследствие меньшей активности брома достигает высоких значений:
Бромирование протекает аналогично реакции с участием простейших алканов с преимущественным замещением водорода у вторичных атомов:
Продуктом этой реакции является 2-бромпропан.
Галогенирование бутана
В реакциях бутана с хлором селективность не играет заметной роли. Даже при низкой температуре соотношение продуктов хлорирования может быть различным:
Бромирование бутана демонстрирует большую селективность:
Реакция отщепления
Активные двухвалентные металлы (как правило, магний или цинк) отщепляют от молекул дигалогеналканов атомы хлора или брома, если они замещают водород у двух соседних атомов углерода. Между последними образуется двойная связь. Продуктом такой реакции является алкен.
Пример реакции дегалогенирования алканов – отщепление цинком атомов хлора от 1,2-дихлорпропана с образованием пропилена (пропена) и хлорида цинка:
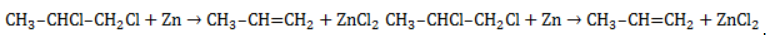
Применение галогеналканов
Хлорированные и бромированные алканы применяются в качестве промежуточных соединений в различных отраслях, таких как синтез высокомолекулярных соединений, производство лаков, красок и растворителей. Хлоралканы служат сырьем для фторалканов, которые нельзя получить прямым фторированием.
Токсичность галогеналканов тем меньше, чем активнее входящий в их состав галоген. Поэтому фторалканы наиболее безопасны. Фторсодержащие фреоны широко используются в качестве вспенивателей, хладагентов и пропеллентов.
Пропан: способы получения и химические свойства
Пропан C3H8 – это предельный углеводород, содержащий три атома углерода в углеродной цепи. Бесцветный газ без вкуса и запаха, нерастворим в воде и не смешивается с ней.
Гомологический ряд пропана
Все алканы — вещества, схожие по физическим и химическим свойствам, и отличающиеся на одну или несколько групп –СН2– друг от друга. Такие вещества называются гомологами, а ряд веществ, являющихся гомологами, называют гомологическим рядом.
Самый первый представитель гомологического ряда алканов – метан CH4. , или Н–СH2–H.
Продолжить гомологический ряд можно, последовательно добавляя группу –СН2– в углеводородную цепь алкана.
| Название алкана | Формула алкана |
| Метан | CH4 |
| Этан | C2H6 |
| Пропан | C3H8 |
| Бутан | C4H10 |
| Пентан | C5H12 |
| Гексан | C6H14 |
| Гептан | C7H16 |
| Октан | C8H18 |
| Нонан | C9H20 |
| Декан | C10H22 |
Общая формула гомологического ряда алканов CnH2n+2.
Первые четыре члена гомологического ряда алканов – газы, C5–C17 – жидкости, начиная с C18 – твердые вещества.
Строение пропана
В молекулах алканов встречаются химические связи C–H и С–С.
Связь C–H ковалентная слабополярная, связь С–С – ковалентная неполярная. Это одинарные σ-связи. Атомы углерода в алканах образуют по четыре σ-связи. Следовательно, гибридизация атомов углерода в молекулах алканов – sp 3 :

При образовании связи С–С происходит перекрывание sp 3 -гибридных орбиталей атомов углерода:

При образовании связи С–H происходит перекрывание sp 3 -гибридной орбитали атома углерода и s-орбитали атома водорода:

Четыре sp 3 -гибридные орбитали атома углерода взаимно отталкиваются, и располагаются в пространстве так, чтобы угол между орбиталями был максимально возможным.
Поэтому четыре гибридные орбитали углерода в алканах направлены в пространстве под углом 109 о 28′ друг к другу:

Это соответствует тетраэдрическому строению.
Например, в молекуле пропана C3H8 атомы водорода располагаются в пространстве в вершинах тетраэдров, центрами которых являются атомы углерода. При этом углеродный скелет образует угол, т.е. геометрия молекулы — уголковая или V-образная.

Изомерия пропана
Для пропана не характерно наличие изомеров – ни структурных (изомерия углеродного скелета, положения заместителей), ни пространственных.
Химические свойства пропана
Пропан – предельный углеводород, поэтому он не может вступать в реакции присоединения.
Для пропана характерны реакции:
Разрыв слабо-полярных связей С – Н протекает только по гомолитическому механизму с образованием свободных радикалов.
Поэтому для пропана характерны радикальные реакции.
Пропан устойчив к действию сильных окислителей (KMnO4, K2Cr2O7 и др.), не реагирует с концентрированными кислотами, щелочами, бромной водой.
1. Реакции замещения
В молекулах алканов связи С–Н более доступны для атаки другими частицами, чем менее прочные связи С–С.
1.1. Галогенирование
Пропан реагирует с хлором и бромом на свету или при нагревании.
При хлорировании пропана образуется смесь хлорпроизводных.
Бромирование протекает более медленно и избирательно.
С третичный–Н > С вторичный–Н > С первичный–Н
Хлорпропан может взаимодействовать с хлором и дальше с образованием дихлорпропана, трихлорпропана, тетрахлорпропана и т.д.
1.2. Нитрование пропана
Пропан взаимодействует с разбавленной азотной кислотой по радикальному механизму, при нагревании и под давлением. Атом водорода в пропане замещается на нитрогруппу NO2.
2. Дегидрирование пропана
Дегидрирование – это реакция отщепления атомов водорода.
В качестве катализаторов дегидрирования используют никель Ni, платину Pt, палладий Pd, оксиды хрома (III), железа (III), цинка и др.
При дегидрировании алканов, содержащих от 2 до 4 атомов углерода в молекуле, разрываются связи С–Н у соседних атомов углерода и образуются двойные и тройные связи.
3. Окисление пропана
Пропан – слабополярное соединение, поэтому при обычных условиях он не окисляется даже сильными окислителями (перманганат калия, хромат или дихромат калия и др.).
3.1. Полное окисление – горение
Пропан горит с образованием углекислого газа и воды. Реакция горения пропана сопровождается выделением большого количества теплоты.
Уравнение сгорания алканов в общем виде:
При горении пропана в недостатке кислорода может образоваться угарный газ СО или сажа С.
Получение пропана
1. Взаимодействие галогеналканов с металлическим натрием (реакция Вюрца)
Это один из лабораторных способов получения алканов. При этом происходит удвоение углеродного скелета.
При проведении синтеза со смесью разных галогеналканов образуется смесь разных алканов.
2. Декарбоксилирование солей карбоновых кислот (реакция Дюма)
Реакция Дюма — это взаимодействие солей карбоновых кислот с щелочами при сплавлении.
R–COONa + NaOH → R–H + Na2CO3
Декарбоксилирование — это отщепление (элиминирование) молекулы углекислого газа из карбоксильной группы (-COOH) или органической кислоты или карбоксилатной группы (-COOMe) соли органической кислоты.
При взаимодействии бутаноата натрия с гидроксидом натрия при сплавлении образуются пропан и карбонат натрия:
CH3–CH2 – CH2 –COONa + NaOH → CH3–CH2 – CH3 + Na2CO3
3. Гидрирование алкенов и алкинов
Пропан можно получить из пропилена или припина:

При гидрировании пропена образуется пропан:

При полном гидрировании пропина также образуется пропан:

4. Синтез Фишера-Тропша
Из синтез-газа (смесь угарного газа и водорода) при определенных условиях (катализатор, температура и давление) можно получить различные углеводороды:
Это промышленный процесс получения алканов.
Из угарного газа и водорода можно получить пропан:
5. Получение пропана в промышленности
В промышленности пропан получают из нефти, каменного угля, природного и попутного газа . При переработке нефти используют ректификацию, крекинг и другие способы.
Химия пропана: технологии преобразования газа
Пропан (C3H8) — органическое вещество класса алканов. Содержится в природном газе, образуется при крекинге нефтепродуктов (высокотемпературная переработка нефти и ее фракций с целью получения, как правило, продуктов меньшей молекулярной массы — моторных топлив, смазочных масел и т. п., а также сырья для химической и нефтехимической промышленности).
Производство бытовых сжиженных газов
Благодаря своим свойствам, таким как высокая теплотворная способность при сгорании, сгорание без остатка, безвредность и безопасность при правильной эксплуатации, удобство в использовании, пропан является универсальным газом и широко используется и на производстве, и в быту.
На сегодняшний день спрос на пропан-бутан огромен. В соответствии с ГОСТ 20448-90, распространяющимся на сжиженные углеводородные газы, предназначенные в качестве топлива для коммунально-бытового потребления и других целей, существуют основные марки сжиженных газов:
- ПТ — пропан технический;
- СПБТ — смесь пропана и бутана технических;
- БТ — бутан технический;
В марках ПТ, СПБТ и БТ содержание метана, этана и этилена не нормируется; пропана и пропилена в ПТ содержится не менее 75 %, а в СПБТ и БТ — не нормируется; содержание бутанов и бутиленов в ПТ не нормируется, в СПБТ их не более 60 %; в БТ их содержится не менее 60 %. Жидкий остаток углеводородов (С5 и выше) составляет не более 1-2 % от объема.
Хлорирование пропана – промышленный метод получения перхлорэтилена
Термическое хлорирование пропана (250-350 °С) приводит к трудноразделяемой смеси моно-и дихлорпропанов, при повышении температуры до 400-500 °С образуются хлорпропены; исчерпывающее хлорирование в избытке хлора при 550-600 °С — один из промышленных методов получения перхлорэтилена и СС14.
Термическое хлорирование пропана в промышленности проводится главным образом с целью производства 1,3-дихлорпропана, на основе которого получается циклопропан. Механизм хлорирования пропана включает следующие стадии: пропан и хлор нагревают раздельно в жидком виде до 400—600°, после чего в поток пропана с большой скоростью вводится хлор с таким расчетом, чтобы скорость его ввода была выше скорости распространения пламени. Реакция проводится в трубчатом змеевике. Так же как и при хлорировании метана, применяется ступенчатая подача хлора с таким расчетом, чтобы на отрезке реакционной трубы между предыдущей и последующей подачей хлора реакция успевала полностью завершиться. Съем избыточного тепла реакции достигается введением с пропаном инертного разбавителя, например, азота или двуокиси углерода. На некоторых установках реакционный змеевик с этой целью помещают в баню с расплавленными солями. Продукты реакции охлаждаются в змеевиковом холодильнике, после чего поступают в ректификационную колонну на разделение. Выделяемые углеводороды вновь направляются на реакцию, а хлорированные углеводороды подвергаются повторной ректификации для разделения на моно-, ди- и полихлориды. Разгонка осуществляется на нескольких колоннах.
Дегидрирование пропана – способ получения пропилена
Дегидрирование пропана как промышленный способ получения пропилена используется с 1990 года. В процессе дегидрирования практически отсутствуют побочные продукты.
В соответствии с данной технологией пропан (и небольшое количество водорода для снижения коксообразования) подают в реактор с неподвижным либо движущимся слоем катализатора при температуре 510-700 ºС при атмосферном давлении. Катализатором служит платина, нанесенная на активированный оксид алюминия, содержащий 20% хрома. При любой конструкции реактора необходима постоянная регенерация катализатора для сохранения его активности. Выходящий из реактора поток поступает в стандартные колонны для разделения. Непрореагировавший пропан и некоторое количество водорода возвращаются в процесс, смешиваясь со свежей порцией сырья. Оставшийся продукт содержит примерно 85% пропилена, 4% водорода, а также легкие и тяжелые отходящие газы.
Применение данной технологии оправдано при высоком спросе на пропилен, превышающем спрос на этилен. Отсутствие побочных продуктов избавляет от дополнительных усилий по их реализации. Одним из ключевых моментов для производства пропилена дегидрированием пропана является разница цен пропилена и пропана. Если разница будет недостаточной, то может оказаться, что производимый пропилен будет стоить дороже, чем по рыночным расценкам. Однако нельзя сказать, что процесс дегидрирования используется лишь при наличии источника достаточно дешевого пропана. Фактически, большинство заводов по дегидрированию пропана расположено в местах, где существует особая потребность в пропилене, а не там, где есть дешевый пропан. В то время как большая часть пропилена производится при переработке нефти и ее продуктов, получение пропилена дегидрированием пропана позволяет получать сырье, которое не связано напрямую с ценами на нефть.
http://chemege.ru/propan/
http://td-np.ru/himiya_propana-html/
Хлорирование (химич.)
Хлорирование органических соединений, процесс прямого замещения в органических соединениях атомов водорода атомами хлора. Хлорирование может быть осуществлено действием свободного хлора или веществами, его генерирующими, например хлористым сульфурилом SO2Cl2 (см. Сульфурила галогениды). Механизм хлорирования определяется природой органического соединения и условиями реакции. Так, насыщенные углеводороды взаимодействуют с хлором при облучении ультрафиолетовым светом (УФ-облучении) по радикально-цепному механизму:
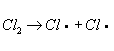 ;
; 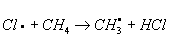 ;
;
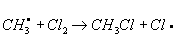 и т. д.
и т. д.
Эта реакция лежит в основе промышленного способа получения из метана метилхлорида, метиленхдорида, хлороформа, четырёххлористого углерода, из пентановых фракций бензина — амилхлоридов. Хлорирование органических соединений ароматического ряда протекает по ионному механизму в присутствии кислотного катализатора, например AlCl3 или FeCl3. Т. о. в промышленности получают, например, хлорбензол:
Cl2 + FeCl3 ® С+ + [FeCl4]—;
C6H6 + Cl+ ® C6H5Cl + Н+;
[FeCl4]— + H+ ® FeCl3 + HCl.
Принимая во внимание различия в механизмах хлорирования органических соединений алифатических и ароматических рядов, регулируют хлорирование жирно-ароматических углеводородов: прибавление FeCl3 ведёт к замещению атомов водорода в ароматическом ядре, тогда как УФ-облучение и повышение температуры способствуют хлорированию боковых алифатических групп. Так, в промышленности хлорированием толуола получают хлортолуолы (в присутствии FeCl3) или бензилхлорид C6H5CH2Cl (под действием УФ-облучения). При высокой температуре удаётся осуществить прямое замещение атомов водорода на хлор и в алкильных группах олефинов (с сохранением кратной связи), например:
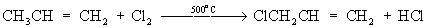
Эта реакция используется в промышленности для получения аллилхлорида — исходного продукта в производстве глицерина.
Иногда под хлорированием в более широком смысле понимают создание связи С—Cl любым способом, например присоединением по кратным связям хлора, хлористого водорода, хлорноватистой кислоты, хлористого нитрозила, замещением на хлор др. функциональных групп (гидроксильной в спиртах и карбоновых в кислотах, аминогруппы в ароматических аминах после предварительного их диазотирования и др.). Так, в промышленности присоединением хлора к этилену получают дихлорэтан, являющийся сырьём в одном из способов производства винилхлорида; хлорированием ацетилена — тетрахлорэтан, применяемый для получения трихлорэтилена, хлорированием некоторых каучуков — хлор-каучуки. Реакцией ненасыщенных соединений с хлористым водородом в промышленности производят винилхлорид, этилхлорид, хлоропрен. Хлорирование используется также для получения инсектицидов (гексахлорана, полихлорпинена, полихлоркамфена), гербицидов, например эфиров 2,4-дихлор-феноксиуксусной кислоты, гексахлорэтана (заменителя камфары) и др. важных продуктов.
Thus, initial chlorination reactions mediated by a transition-metal catalyst were performed on β-keto esters in the presence of 5mol % of [TiCl2((R,R)-TADDOLato)] catalysts 7 or 8 with N-chlorosuccinimide (NCS) in analogy to electrophilic fluorination reactions.
From: Comprehensive Chirality, 2012
Synthesis: Carbon With One Heteroatom Attached by a Single Bond
S.P. Stanforth, in Comprehensive Organic Functional Group Transformations II, 2005
2.12.3.2.1 Aryl chlorides by electrophilic aromatic substitution
Several examples of electrophilic chlorination reactions that use either HCl or KCl in the presence of an appropriate oxidizing agent have already been discussed in the context of general methods for halides (Section 2.12.1.2.1). KCl and oxone™ have also been used for the electrophilic chlorination of toluene, acetanilide, and phenol in acetonitrile at room temperature <2002SC279>. High conversions were achieved under these conditions and mixtures of ortho— and para-substituted products were obtained, for example chlorination of toluene gave a 70:30 ratio of 4-chlorotoluene and 2-chlorotoluene. Chlorobenzene, nitrobenzene, and benzoic acid were unreactive even when the reactions were carried out at 80 °C. Aromatic compounds have been chlorinated in good yield under mild conditions using a mixture of tin tetrachloride and lead tetraacetate in dichloromethane <1996T8863>. For example, toluene gave a 45:55 ratio of 4-chlorotoluene and 2-chlorotoluene in 80% yield. Bromobenzene and iodobenzene could also be chlorinated giving 4-bromochlorobenzene and 4-chloroiodobenzene in yields of 56% and 58%, respectively.
t-Butyl hypochlorite in the presence of zeolites has been used as an efficient system for the monochlorination of a wide variety of mono- and di-substituted aromatic substrates under mild conditions <1999GC83>. The best system was generally t-butyl hypochlorite with HNaX in acetonitrile, which allowed high yielding (75–100%) monochlorinations of benzene, alkylbenzenes, anisole, chlorobenzene, and bromobenzene. Mixtures of ortho— and para-substituted products were obtained with the best selectivity being obtained for t-butylbenzene (para:ortho = 98:2) and the worst for anisole and toluene (para:ortho = 82:12 for both compounds).
Sodium chlorite has been used as a chlorinating reagent on several occasions. Thus, in combination with trichloroacetic acid (which generates chlorous acid, HOClO, in situ) benzene gave chlorobenzene in low yield (20%) and mesitylene underwent monochlorination in 95% yield <1999M1493>. Toluene, however, gave a mixture of 4-chlorotoluene (50%), 2-chlorotoluene (30%), and benzyl chloride (20%). In combination with the (salen)manganese(III) complex 4 and moist alumina, sodium chlorite has been used to chlorinate alkyl phenyl ethers in good yield with excellent para-regioselectivity in dichloromethane at room temperature <1997CJC1905>. Mesitylene also underwent monochlorination under these conditions but o-, m-, and p-xylenes were unreactive. Manganese(III) acetylacetonate, moist alumina, and sodium chlorite have also been used for the chlorination of alkyl phenyl ethers with high para-selectivity <1997JCS(P1)3081>.

Aryl chlorides have been produced from the reaction of substituted benzene derivatives and acetyl chloride in the presence of a catalytic quantity of ceric ammonium nitrate (CAN) (Equation (38)) <2003SL221>. The reaction proceeds in acetonitrile at room temperature but is restricted to activated substrates. Acetyl chloride in combination with manganese(III) acetate has also been used for the chlorination of activated substrates and this reaction is significantly accelerated when performed under sonication <1996JCR(S)164>.
(38)
Sulfuryl chloride has often been used as a chlorinating agent for electron-rich aromatic compounds and its use has been extended to include the chlorination of aryl alkylamines, without need for protection of the amine group (Equation (39)) <2001TL3247>. For the five examples reported, yields were good (60–80%). Merrifield resin bound o-cresol underwent chlorination with sulfuryl chloride giving para:ortho ratios in excess of 50:1 <2002T8059>.
(39)
Phenol and o-cresol have both been chlorinated by N-chloro-2,3,4,4,5,6-hexachlorocyclohexa-2,5-dienylamine 5 in a reaction in which the ortho/para selectivity was solvent dependent, as illustrated for the chlorination of phenol in (Equation (40)) <2002SC735>.

(40)
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0080446558000349
The Integration of Process Design and Control
Hasan Y. Alhammadi, Jose A. Romagnoli, in Computer Aided Chemical Engineering, 2004
Step 3: Establish energy management system
The direct chlorination and oxy-chlorination reactions are exothermic reactions and good temperature controllers are required to keep the reactions at the optimum conditions. For the case of no heat integration within the plant processes, the temperatures of the reactors are controlled by the flow rate of the cooling water streams. The temperature of the endothermic cracking reaction is controlled by the fuel gas, heating utility, to be kept at the optimum temperature. For the quench processes, cooling water is used first to cool down the hot reactor streams before they proceed to the refrigeration sections, if required, so that the refrigeration load is reduced.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/S1570794604800634
Synthetic Methods V – Organocatalysis
S. Kotani, M. Nakajima, in Comprehensive Chirality, 2012
6.21.3 Mechanistic Scope of the Chlorination Reaction by Silicon Tetrachloride and Chiral Lewis Bases
Although several Lewis base-catalyzed chlorination reactions of meso epoxides have been developed, attempts to elucidate the detailed reaction mechanism are ongoing.
Denmark’s group, a pioneer in terms of Lewis base-catalyzed reactions involving hypervalent silicates, has extensively discussed the ring-opening mechanism using chiral phosphoramides as organocatalysts. A simple complex involving a hypervalent silicate generated from silicon tetrachloride and a chiral phosphoramide was proposed in their first publication (Figure 17).13 In this mechanism, a complex generated by the coordination of the silicon tetrachloride and the phosphoramide is ionized to produce a highly reactive silylium cation and a nucleophilic chloride anion. The addition of a chloride ion to the epoxide which is activated by a chiral silylium cation/phosphoroamide complex occurs in an SN2 fashion via a pentacoordinate silicate complex intermediate.

Figure 17. Proposed reaction mechanism promoted by the complex of phosphoramide and SiCl4.
Fu and coworkers explored the mechanism of the enantioselective ring-opening process using their original planar-chiral pyridine N-oxide and made several interesting observations.14 A positive nonlinear relationship was observed between the ee of the catalyst and the ee of the product. This observation suggests the possibility of the coordination of the second pyridine N-oxide site to silicon atom forming a hexacoordinate hypervalent silicate.
Nakajima and coworkers designed and synthesized pyridine N-monooxide, wherein one of the two binding sites of an N-oxide catalyst was substituted with a methyl or methoxy group. They examined the ring opening of the cis-stilbene oxide.15 Because the catalytic activities of both N-monooxides were quite low compared to that of the bipyridine N,N′-dioxide, no products were obtained at −78 °C for the cis-stilbene oxide. They then used 1,4-bisbenzyloxy-2-butene oxide as a substrate, which gave the corresponding chlorohydrin from the pyridine N-monooxide derivatives, but low chemical and optical yields were observed with both pyridine N-monooxides (Figure 18). The significant decrease in reactivity and selectivity could be explained by the hypothesis that pentacoordinate silicate formed by the coordination of N-monooxide was less reactive and selective, producing a less selective product, whereas the bidentate catalyst provided a highly reactive hexacoordinate intermediate, which allowed the products to give high yields and enantioselectivities (Figure 19).

Figure 18. Reactivity and selectivity relationship of the pyridine N-oxide catalyst in the reaction of 1,4-dibenzyloxy-2-butene oxide.

Figure 19. Proposed reaction mechanism promoted by the complex between the bipyridine N,N-dioxide and SiCl4.
The additive was crucial for both the reactivity and selectivity. The addition of diisopropylethylamine was found to increase the rate of reaction in the Lewis base-catalyzed allylation of aldehydes by allyltrichlorosilane.25 Fu and Nakajima applied diisopropylethylamine as an additive to the chlorination of meso epoxides by silicon tetrachloride, providing the corresponding chlorohydrins in high yields and enantioselectivities.14,15 Diisopropylethylamine works as a scavenger of hydrogen chloride, which is produced through the adventitious hydrolysis of the chlorosilyl reagents. The amine traps hydrogen chloride, preventing epoxide from reacting directly to form chlorohydrin in a nonstereoselective process. As a result, both yields of the corresponding products were reproducible.
Denmark and coworkers found that the hydrogen chloride generated from the silyl chlorides in the quenching reaction rapidly opened the unreacted epoxide to afford the cyanohydrin nonselectively.8 If the reaction did not proceed to completion, the racemate generated from the unreacted epoxide with hydrogen chloride led to erroneous conversion and selectivity. After a considerable effort toward the screening of quenching methods, they found that the addition of propylene oxide, which consumed the unreacted silyl reagents before quenching, avoided formation of the racemic chlorohydrin. They also examined the 29Si NMR spectra, which have proven to be the tool of choice for the detection of silyl cations. Their studies involving silicon tetrachloride and HMPA showed three majors signals. The signal at −19 ppm was assigned to the free silicon tetrachloride, and two other signals appeared at −110 and −206 ppm (Table 1). The signal at −110 ppm corresponded to the bis-HMPA complex of the trichlorosilyl cations. The signal that was shifted to the furthest upfield (−206 ppm) was assigned to the hexachlorosilicate (SiCl62−). 31P NMR analysis indicated that a HMPA complex with silicon tetrachloride appeared at 19 ppm, accompanied by the concomitant disappearance of the free HMPA signal at 27 ppm. These results suggested that a complex of the phosphoramide and silicon tetrachloride was the active species in the halogenation of the epoxides.
Table 1. 29Si and 31P NMR analysis of the reaction between silicon tetrachloride and HMPA
The positive nonlinear relationship observed in the presence of the pyridine N-oxide catalyst supported a positive influence of the bidentate catalysts on the stereodetermining step.14 Nakajima’s results, shown in Figure 17, also supported the importance of two basic oxygen atoms bound to the silicon atom. Their monodentate N-oxide catalysts most likely had difficulty forming a complex with silicon tetrachloride due to the steric hindrance of the catalysts. Consequently, both the reactivity and selectivity dramatically decreased.
Denmark and coworkers reported a negative nonlinear effect between the ee of the phosphoramide catalyst and the ee of product.8 Furthermore, 29Si and 31P NMR analysis suggested the presence of a complex between the Lewis base catalyst and silicon tetrachloride. These observations supported the hypothesis that two sites were required to form a coordination complex with the silicon in the transition structure. They examined the halogenation of cis-stilbene oxide using dimeric phosphoroamide catalysts. However, none of the bidentate catalysts gave enantioselectivities that were better than those of the monophosphoramide catalyst (Table 2). These results did not eliminate the possibility that two binding sites were involved in the stereodetermining transition structure, suggesting that these sites were not in close proximity. Because the strength of the basicity of catalyst and the activity of the silyl complex formed with silicon tetrachloride depends on the structure of the Lewis base, the transition states may be fundamentally not common.
Table 2. Enantioselective chlorination of cis-stilbene oxide catalyzed by phosphoramides
On the basis of 29Si NMR analysis and the catalyst structure survey, Denmark proposed a second mechanism (Figure 20).8 In this mechanism, the silicon complex derived from silicon tetrachloride and phosphoramide undergoes ionization to form a reactive silylium cation and a chloride anion, which forms a complex with another silicon complex to afford a highly nucleophilic hexacoordinate silylium anion. The chloride ion in the chiral silicon complex attacks the meso epoxide to give the corresponding chlorohydrin.

Figure 20. The second plausible mechanism promoted by the complexes between phosphoramide and SiCl4.
Various types of Lewis base organocatalysts have been designed and applied to the chlorination of meso epoxide with silicon tetrachloride. This process provides an efficient system for the preparation of enantiomerically pure chlorohydrin. Further investigations that clarify the detailed mechanism and improve the generality of the substrates are ongoing and are under debate.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780080951676006248
Synthetic Methods IV – Asymmetric Oxidation Reduction, C–N
N. Shibata, … D. Cahard, in Comprehensive Chirality, 2012
5.9.3.2 Transition-metal Catalysis
With rare exceptions, all the catalysts used in chlorination reactions were previously developed for fluorination reactions. Most of the substrates were β-keto esters but we have also seen chlorination of a β-diketone, oxindoles, imides, and some β-keto phosphonates. Various chlorine donors were evaluated (Figure 4) and the results of enantioselective electrophilic chlorination on β-keto esters are listed in Table 1. Already described catalysts and ligands as well as new ones are depicted in Figure 5.

Figure 4. Electrophilic chlorine donors.
Table 1. Enantioselective electrophilic chlorination on β-keto esters catalyzed by transition-metal complexes
| Entry | Substrates | Conditions | Cl-donor | Yield (%) | ee (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Catalyst 7 or 8 (5 mol%) MeCN, r.t., 30 min–15 h | NCS | 85–97 | 11–88 | 2000 HCA 2425 | |
| 2 | Catalyst 7 (5 mol%) Pyridine (1.2 equivalent) Toluene, 50 °C, 20 min | 37–83 | <10–71 | 2001 HCA 605 | ||
| 3 | (S, S)-t-Bu-Box 27 (10 mol%) Cu(OTf)2 (10 mol%) Et2O, r.t., 1 h | NCS | 88–99 | 48–77 | 2001 CEJ 2133 | |
| 4 | (R, R)-Ph-DBFOX 15 (0.11 equivalent) Ni(ClO4)2·6H2O (0.1 equivalent) CH2Cl2, MS, r.t., 3–45 h | CF3SO2Cl | 66–85 | 94–98 | 2005 ACIE 4204 | |
| 5 | Ligand 18 (5 mol%) Ni(ClO4)2 (5 mol%) CH2Cl2, MS, r.t., 2 h | 99 | 92 | 2008 CL 1098 | ||
| 6 | Ligand 23 (10 mol%) Cu(OTf)2 (10 mol%) Et2O, −78 °C to r.t., 16 h | NCS | 88–99 | 24–91 | 2009 EJOC 4085 | |
| 7 | Ligand 20 (10 mol%) Co(acac)2 (10 mol%) Toluene, r.t., MS, 12 h | CF3SO2Cl | 62 | 88 | 2010 CL 466 | |
| 8 | Ligand 28 (5 mol%) CuOTf.1/2 C6H6 (5 mol%) CH2Cl2, 0 °C, 2 h | NCS | 95–99 | 47–83 | 2010 TA 247 |

Figure 5. Catalysts and ligands used in chlorination reactions.
Pioneering work on transition-metal catalysed enantioselective electrophilic chlorination was conducted after observing the incorporation of chlorine ligand into β-keto esters during asymmetric fluorination in the presence of [TiCl2((R,R)-TADDOLato)] catalysts. Thus, initial chlorination reactions mediated by a transition-metal catalyst were performed on β-keto esters in the presence of 5 mol % of [TiCl2((R,R)-TADDOLato)] catalysts 7 or 8 with N-chlorosuccinimide (NCS) in analogy to electrophilic fluorination reactions. The bulkier catalyst 7 tended to give higher ee values in shorter reaction times with the best result being obtained in the chlorination of a bulky diphenylmethyl acyclic β-keto ester (entry 1, Table 1).21 Dichloro(4-methylphenyl)iodine is also a suitable chlorinating agent in the chlorination of β-keto esters albeit with slightly lower yields and ee values (entry 2, Table 1).22 Chiral bisoxazoline copper(II) complexes such as (S,S)-t-Bu BOX 27 / Cu(OTf)2 gave optically active α-chloro-β-keto esters in high yields and moderate enantioselectivities (entry 3, Table 1).23 In this case, the evaluation of other chlorinating agents A–D (see Figure 4) resulted in lower enantioselectivities relative to NCS whereas additives such as bases or HFIP did not improve the ee. Much higher enantioselectivities were attained with the aid of Ni(II)/(R,R)-Ph-DBFOX 15 for chlorination of cyclic β-keto esters derived from indanone and tetralone (entry 4, Table 1).12 Importantly, the use of CF3SO2Cl as a chlorinating agent is essential to achieve high enantioselectivity, since NCS provided a significant drop in ee. A single example of chlorination was reported with the chiral tridentate ligand 18/Ni(ClO4)2 combination leading to a high ee only by slow addition of the chlorinating agent, perchloro-2,4-cyclohexadien-1-one D, to the reaction mixture containing the substrate and the catalyst; changing the operation sequence led to the racemic chlorinated product (entry 5, Table 1).14 C1-symmetric amino sulfoximine–copper complex 23 has been used as chiral catalyst in chlorination reactions of β-keto esters in the presence of NCS as a source of electrophilic chlorine (entry 6, Table 1).17 The best result (91% ee) was obtained in the chlorination of ethylcyclohexanone-2-carboxylate, a substrate that regularly gives poor stereodifferentiation; however, the analog substrate having a cyclopentanone ring only gave 39% ee. t-Butylcyclopentanone-2-carboxylate was chlorinated in up to 88% ee with CF3SO2Cl in the presence of (R,R)-Jacobsen’s salen ligand 20 in combination with Co(acac)2 (entry 7, Table 1). Molecular sieves were required to increase both the yield and enantioselectivity.15 Finally, the chiral oxazoline-Schiff base ligand 28 was developed only for chorination. Over the metal employed, copper(I) triflate. ½ C6H6 provided high catalytic activity for the chlorination conducted with NCS (entry 8, Table 1).24
It is evident that the choice of the ligand/metal combination as well as the source of chlorine donor is essential for achieving high enantioselectivities. The type of substrate (acylic, cyclic, with or without fused aromatic ring, bulkiness of the ester moiety) is obviously also important. Nevertheless, the availability or simplicity to prepare the chiral ligand will certainly orientate the choice when such chlorination is required in a synthetic plan.
Apart from β-keto esters, one case of chlorination of a 1,3-diketone was reported with the aid of (S,S)-t-Bu BOX 27/Cu(OTf)2 giving optically active α-chloro derivative in 99% yield with only 32% ee (equation 40).23 Oxindoles have been chlorinated with CF3SO2Cl and the Ni(II)/(R,R)-Ph-DBFOX 15 combination with up to 96% ee; one of them was converted into BMS-225113, a pharmaceutically active chlorooxindole (equation 41).12 In addition, the enantioselective chlorination of phenyl acetylthiazolidinone was achieved with the Ni(II)/(R,R)-Ph-DBFOX 15/HFIP/2,6-lutidine combination in high yield and excellent enantioselectivity (equation 42).11
(40)
(41)
(42)
Acyclic and cyclic β-keto phosphonates were readily chlorinated to α-chloroalkylphosphonates in a similar way to fluorination. In a comparison of the two halogenations, chlorination versus fluorination, the use of NCS allowed slightly better ee values than NFSI. The best combination was Zn(SbF6)2/(R,R)-Ph-DBFOX 15 in dichloromethane at room temperature (equation 43).9
(43)
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780080951676005115
Nonenvironmental Industrial Applications of Activated Carbon Adsorption
Meenakshi Goyal, in Novel Carbon Adsorbents, 2012
20.12.6 Combination Reactions
Activated carbons have also been used as catalysts for several combination, decomposition, elimination, and chlorination reactions. The combination of hydrogen and bromine has been carried out using several activated carbons having different surface areas and associated with varying amounts of the carbon–oxygen surface groups. The reaction efficiency was found to be between 80 and 100% at a temperature between 80 and 150 °C in the presence of the activated carbon [116,117]. The reaction did not take place in the absence of the carbon. The catalytic activity was related to the surface area of the carbon while it was retarded by the presence of carbon–oxygen surface groups on the carbon surface. It has been suggested [118,119] that both hydrogen and bromine are adsorbed on the carbon surface at the neighboring sites. The activated carbon surface probably activates the bromine molecule in a manner similar to its activation by light. The activated bromine then reacts with the hydrogen to give HBr which is desorbed, making the active sites available and starting a chain reaction.
The combination of phosgene and formaldehyde for the formation of dichloromethane in the temperature range 180–200 °C is also catalyzed by activated carbons [120]. There is almost complete conversion of the reactants below 200 °C in the presence of activated carbons. However, when the reaction is carried out above 200 °C, the percentage conversion into dichloromethane is considerably decreased. Temperature-controlled desorption studies indicated that both phosgene and formaldehyde were adsorbed on the carbon surface and then reacted to produce an intermediate compound which then decomposed into dichloromethane and CO2.

Several decomposition reactions are catalyzed by activated carbons. The decomposition of H2O2 has been studied as a function of the concentration of the solution, the temperature, as well as the nature of the carbon surface. The decomposition increases with concentration of the solution and the temperature. The catalytic activity of a carbon is considerably enhanced in the presence of degassed carbon samples. In fact, the carbons which are associated with carbon–oxygen surface groups inhibit the decomposition of H2O2 [121–123]. The results show that although the acidic medium is conducive to the stability of H2O2, an alkaline medium is conducive to its decomposition. The function of the carbon is simply to provide an environment of a particular degree of acidity or alkalinity. Thus, while acidic activated carbons inhibit the decomposition, the basic carbons enhance the decomposition. The decomposition has been attributed to the existence of chromene groups [124] which initiate a chain reaction for the decomposition. At low pH values, these chromene groups are oxidized to carbonium ions and thus the carbon loses its ability to decompose H2O2.
The catalytic decomposition of benzoyl peroxide in organic solutions has been investigated by several workers. The decomposition in benzene solutions increases considerably with concentration of the solution, the temperature, and the amount of the carbon used. The decomposition produces benzoic acid and CO2. The amount of benzoyl peroxide decomposed is related linearly to the surface area of the activated carbon. The decomposition activity of the carbon catalyst is considerably reduced when the same carbon was reused in a second cycle. This is attributed to the fact that some of the carbon surface has been covered by the chemisorbed free radical benzoyl species C6H5COO−.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B978008097744700020X
Six-membered Rings with Two or More Heteroatoms and Fused Carbocyclic Derivatives
William J. Coates, in Comprehensive Heterocyclic Chemistry II, 1996
6.01.7.7 Other Substituted Alkyl Groups
The reactions of other substituted alkyl groups were not specifically addressed in CHEC-I except for oxidation and chlorination reactions of hydroxyalkyl groups 〈84CHEC-I(3B)1〉. α-(3-Pyridazinyl)phenylmethanol is easily converted with thionyl chloride into the chloride (Scheme 53) which undergoes successful displacement reactions with a range of amines and sulfur nucleophiles 〈89JHC1787〉. The only products isolated are those from direct displacement of chloride, in contrast to reactions with the 4-pyridazinyl isomer in which some tele substitution also occurs 〈84M1171〉. Chlorination of 3,6-dichloro-4-(2-hydroxy-1,1-dimethylethyl)pyridazine with thionyl chloride gives the corresponding fungicidal chloro compound in 76% yield as the final step in a potential industrial preparation 〈88JOC5704〉.

Scheme 53.
Nitration of 4-hydroxymethyl-1(2H)-phthalazinone and its derivative EG-626 with acetyl nitrate has been shown to give, after formation of the methyl nitrate esters, the N-2-nitro derivatives (Scheme 54). An earlier suggestion that the second nitration of EG-626 occurred on the phthalazinone oxygen was disproved by a combination of spectroscopic methods and an x-ray structure determination 〈92CPB3327〉.

Scheme 54.
Oxidative decyanation of phenyl-3-pyridazinylacetonitriles, by passing a brisk stream of oxygen into an aqueous sodium hydroxide/DMSO solution of the substrate containing triethylbenzylammonium chloride, is a convenient method for the synthesis of phenyl 3-pyridazinyl ketones (Scheme 55). Modified reaction conditions allow the one pot conversion of α-(6-chloropyridazin-3-yl)phenylacetonitrile into ketones with concomitant hydrolysis or methoxylation of the chloro substituent; the direct one pot preparation of 3,6-dibenzoylpyridazine from 3,6-dichloropyridazine is achieved in 48% yield 〈93JHC1685〉.

Scheme 55.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780080965185001179
Azo Dyes
Ghodsi Mohammadi Ziarani, … Hendrik G. Kruger, in Metal-Free Synthetic Organic Dyes, 2018
4.1.6 Synthesis of Pyrazole Azo Dyes
Cui et al. [78] described the synthesis of a yellow pyrazole azo disperse dye 208 starting from p-aminobenzoic acid 11 applying successive diazotization, coupling reaction, chlorination, and esterification reaction with ethanol (Scheme 4.45). The synthesized dye was applied to poly(ethylene terephthalate) fabric and it was found that the ester-containing disperse dye showed good alkali-clear ability on this material. The advantages of this dye include little contamination to the environment because of the absence of reductants, as well as low toxicity and easy recycling of the hydrolysates.

Scheme 4.45.
Synthesis of several pyrazole disperse dye derivatives 211 was developed utilizing a coupling reaction of active methylene compounds 210 with aryl diazonium chloride (prepared from the reaction of sodium nitrite and amino pyrazoles 209) (Scheme 4.46) [79]. It was observed that dye 211 with three methyl groups (R CH3, X = meldrum’s acid as electron-donating substituent) showed a bathochromic effect in its UV–Vis spectrum.
CH3, X = meldrum’s acid as electron-donating substituent) showed a bathochromic effect in its UV–Vis spectrum.

Scheme 4.46.
Preparation of a series of pyrazole derivatives containing azo dye moiety 214 was reported by Dabholkar et al. [80] starting from the reaction of p-chlorobenzaldehyde 31 and ethyl acetoacetate 212 through the formation of several intermediates A–F. In the final step, the intermediate F was treated with azo dye containing 1, 3-diketones 213 to afford pyrazolo azo dye 214 in good yields (Scheme 4.47).

Scheme 4.47.
Several pyrazole dyes were synthesized by El-Kholy and coworkers [81] in a sequential strategy. First, thiosemicarbazides 215 were reacted with acetoacetanilide 216 to form acetoacetanilid thiosemicarbazon derivatives A [82]. The coupling reaction of A with diazonium chloride led to isolation of compounds B, which in the presence of a base afforded the final pyrazole dyes 217 (Scheme 4.48). The color intensities and dye fixation on the fabric were also determined, and the results suggested the dye can be used in practice.

Scheme 4.48.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128156476000042
Electrophilic Aromatic Substitution
Robert J. Ouellette, J. David Rawn, in Organic Chemistry, 2014
Friedel–Crafts Acylation
An acyl group can replace hydrogen in an aromatic ring by a reaction called Friedel–Crafts acylation. The reaction requires an acyl halide and the corresponding aluminum trihalide. The reaction is commonly carried out only with acyl chlorides. The electrophile is shown as an acyl cation, called an acylium ion, forms from a Lewis acid–Lewis base complex of aluminum trichloride and the acyl chloride.

Acyl cations are resonance stabilized. The more stable form has an octet of electrons on both the carbon and oxygen atoms, and a formal positive charge on the oxygen atom. However, to give a stable product, reaction of the acyl cation with an aromatic ring must occur at the acyl carbon atom.
![]()
Acylation of benzene by carbocations, such as the propanoyl cation, occurs by a two-step mechanism similar to the mechanism for alkylation.

Problem 13.4
Draw the structures of all possible products formed by monosubstitution of o-dibromobenzene in a chlorination reaction. Do the same for m— and p-dibromobenzene.
Problem 13.5
Draw the structures of all possible products formed by mononitration of tetralin.

Sample Solution
The molecule has a plane of symmetry perpendicular to the plane of the page that bisects both rings. Two nonequivalent hydrogen atoms lie on either side of the plane. Therefore, only two mononitrated products are possible.

Problem 13.6
Write the structures of the two possible products formed by sulfonation of naphthalene. At 80 °C, isomer I constitutes 96% of the reaction mixture. At 165 °C, isomer II is 85% of the reaction mixture. When isomer I is heated in sulfuric acid at 165 °C, it is converted into isomer II. Explain these observations. Based on steric considerations, which isomer is likely to be more stable?
Problem 13.7
Reaction of 3-phenylpropanoyl chloride with AlCl3 yields a compound with the molecular formula C9H8O. Write the structure of the compound and explain its origin.

Sample Solution
The Friedel–Crafts reaction we have discussed in this section is an intermolecular reaction between an aromatic compound and an acyl halide. However, if the acyl halide also contains an aromatic ring, an intramolecular reaction can easily occur. Such a unimolecular reaction is favored over a bimolecular reaction when the ring formed contains either five or six atoms. This is a Friedel–Crafts reaction in which the acyl chloride acylates the ortho position.

Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128007808000139
Synthesis: Carbon With One Heteroatom Attached by a Single Bond
I. Shcherbakova, A.F. Pozharskii, in Comprehensive Organic Functional Group Transformations II, 2005
2.03.10.1.6 By rearrangement
Chlorination of O,O-dialkyl-N,N-dialkyl thiophosphoroamidates using phosphorus oxychloride proceeds with rearrangement to give S-alkyl-N,N-dialkyl chlorophosphates in good yield (Equation (138)) <1995PS(101)91>. In a similar way, the isomerization-chlorination reaction of O,O-dialkyl thiophosphoramidates has been employed to afford the corresponding 3-alkyl chlorophosphates <1996HAC207>.
(138)
The photoirradiation of thionophosphates in acetonitrile leads to thiolophosphates through an established nonchain radical pathway (Scheme 146) <2001JCS(P1)323>. This behavior of thionophosphates is unlike that of the related phosphates, which react via an ionic dissociation–recombination pathway.

Scheme 146.
Sodium iodide has been found to be an effective reagent to induce the thiono–thiolo rearrangement of O,O-diethyl difluromethylenephosphonothioate (Equation (139)) <2000JOC5858>.
(139)
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0080446558000258
Alkanes and Cycloalkanes
Robert J. Ouellette, J. David Rawn, in Organic Chemistry, 2014
Reactivity and Selectivity
We know that bromine is less reactive than chlorine in the rate-determining step of halogenation of methane. We also recall that bromine is more selective than chlorine. Both the rate of reaction and the selectivity of free radical halogenation are related to the first propagation step, so let’s look at the relationship between reactivity and selectivity in terms of the structure of the transition states for chlorination and bromination.
Because the first propagation step of the chlorination reaction is very slightly endothermic, the transition state is only moderately past the “center” of the reaction coordinate axis. In contrast, the transition state for the more endothermic bromination reaction is farther along the reaction coordinate axis (Figure 4.19b). What does this information tell us about the structure of the transition states for the two reactions? We recall that the Hammond postulate states that the structures of transition states most closely resemble those species that are most similar in energy (Section 3.11. Thus, the structure of the transition state for an exothermic process is more reactant-like, or “early,” and the structure of the transition state for an endothermic process is more product-like or “late.”

Figure 4.19. Potential Energy Diagrams for Halogenation Reactions
The energy of activation for the abstraction of a hydrogen atom by a chlorine atom (a) is smaller than the energy of activation for the abstraction of a hydrogen atom by a bromine atom (b).
The first propagation step for the chlorination reaction is very slightly endothermic. Thus, the position of the transition state along the reaction coordinate is a bit past the midpoint (Figure 4.20). The extent to which the C─H bond is broken and the extent to which the H─Cl bond is formed are comparable because the C─H and H─Cl bond dissociation energies are similar.

Figure 4.20. Transition State Structures for Halogenation Reactions
(a) In the transition state for abstraction of hydrogen by a bromine atom, the C─H bond is mostly broken and the H─Br bond is mostly formed.
(b) In the transition state for abstraction of hydrogen by a chlorine atom, the C─H bond is broken to a lesser degree and the H─Cl bond is only partially formed.
The first propagation step for the bromination of methane is strongly endothermic, so the transition state is more product-like. The C─H bond is broken to a significant extent, and the H─Br bond is more strongly formed (Figure 4.20).
How does the degree of bond breaking in the transition state affect the selectivity of the reagent? In the less reactive bromination reaction, the transition state for the first propagation step is more product-like and resembles the radical product. Thus, the ease with which the C─H bond is broken reflects the effect of alkyl groups on the stability of the radical. The reaction then shows a selectivity that reflects the stability of the radical product. In the chlorination reaction, the transition state is more reactant-like, and the alkyl group has not developed much radical character. Hence, the type of C─H bond—primary, secondary, or tertiary—has little effect on the reaction, and low selectivity is the result.
Throughout our study of organic reactions, we will find that, for structurally similar reactions, the more reactive reagent is less selective. Many factors account for this selectivity, including inductive, resonance, and steric effects. Regardless of the factors that influence the rate, the less reactive reagent has a more fully developed transition state (more like the products), and the role of structural features in stabilizing the transition state becomes more important.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128007808000048

Елена Шаповалова Владимировна
Эксперт по предмету «Химия»
Задать вопрос автору статьи
Насыщенные углеводороды активно вступают в реакцию с хлором. Хлорируют алканы атомами или катионами хлора, которые более реакционноспособны, чем молекулярный хлор. Диссоциация молекулы хлора на атомы требует затраты 242,8 кДж/моль энергии. Такая диссоциация хлора легко происходит при обычной температуре под действием УФ-света, поглощение которого молекулой придает ей 293,0 кДж/моль энергии. Для термической диссоциации молекулы хлора на атомы необходима температура около $300^circ C$ . Диссоциация молекулы хлора на ионы требует затраты 1130,2 кДж / моль. Из приведенных энергетических данных видно, что насыщенные углеводороды легче хлорировать на свете.
Хлорирование алканов происходит с выделением 108,8 кДж / моль теплоты и является менее экзотермическим процессом, чем фторирование. Фотохимическое хлорирование алканов проводят при рассеянном свете, поскольку при прямом освещении реакция происходит со взрывом. При хлорировании атомы водорода алканов постепенно замещаются на хлор. В результате образуются хлоропохидни насыщенных углеводородов.
Итак, если смесь метана с хлором нагреть до $200^circ C$ или воздействовать на неё УФ-светом подходящей длины волны, протекает сильно экзотермическая реакция:
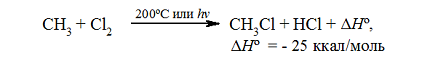
Рисунок 1. Хлорирование метана
Тепловой эффект первой стадии хлорирования метана в газовой фазе до $CH_3Cl$ и $HCl$ может быть рассчитан на основании закона Гесса.
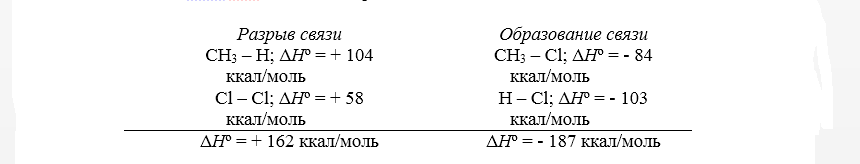
Рисунок 2. Хлорирование метана
Замечание 1
Суммарный тепловой эффект $Delta underline{H}^circ = — 25$ ккал/моль. Эти данные показывают, что хлорирование метана представляет собой вполне вероятный процесс, хотя тепловой эффект никоим образом не связан со скоростью реакции, которая определяется свободной энергией активации.
«Хлорирование метана» 👇
Радикальный механизм хлорирования метана
Хлорирование алканов при нагревании, облучении и в присутствии радикалообразующих инициаторов происходит радикально-цепным механизмом $SR$ (Семенов) и состоит из трех основных стадий:
-
зарождение цепи (инициирование)
Рисунок 3. Хлорирование метана
-
рост цепи
Рисунок 4. Хлорирование метана
-
обрыв цепи (рекомбинация)
Рисунок 5. Хлорирование метана
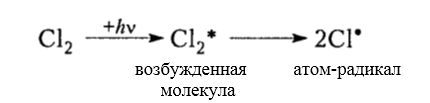
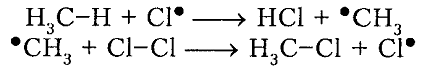
Итак, молекула хлора $Cl_2$ под действием света ($h nu $) или при нагревании получает избыток энергии, становится возбужденной и распадается на два атома, которые по своей природе являются радикалами. Атом-радикал хлора в процессе столкновений или взаимодействия с другими молекулами отщепляет атом водорода от молекулы метана $CH_4$ с образованием метильного радикала $^*CH_3$. Метательный радикал, в свою очередь, отщепляет атом хлора от следующей молекулы $Cl_2$ и т.д. Таким образом, один образованный радикал инициирует много повторений стадии роста цепи. Количество таких повторений определяет длину кинетического цепи всей реакции, для хлорирования может достигать $10 000$ и более.
На рис. 6 показана энергетическая диаграмма хлорирования метана.
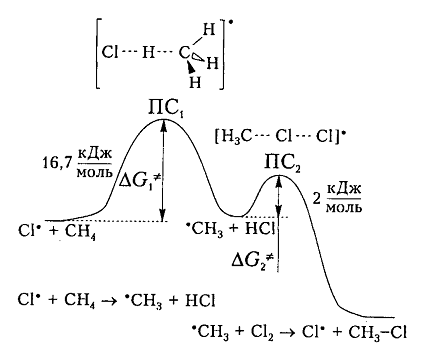
Рисунок 6. Диаграмма изменения свободной энтальпии для взаимодействия атома хлора и молекулы метана
Закономерности радикального хлорирования метана
Исходя из общетеоретических соображений атом хлора и молекула метана будут взаимодействовать между собой только тогда, когда их свободная энтальпия будет равна величине энергетического барьера (и энергии активации) 16,7 кДж / моль, которая всегда немного больше, чем просто разница (-6 кДж / моль) энергий разрыва старой связи $C-H$ (+425 кДж / моль) и образования новой связи — связи $H-Cl$ (-431 кДж / моль). Поэтому не каждое столкновение реагирующих частиц вызывает их взаимодействие, а только те, которые достаточны для преодоления этого барьера.
Дополнительная энергия активации возникает благодаря облучению или нагреву молекул. Возбужденые молекулы проявляют достаточно высокие скорости движения, кинетическая энергия которого и превращается в потенциальную энергию при столкновениях. На вершине кривой в переходном состоянии $ПС_1$ между реагирующими компонентами образуется активированный комплекс, в котором разрыв старого связи $C-H$ и формирования новой ${rm H}-Cl$ происходят примерно одновременно. Образованный метательный радикал имеет достаточную потенциальную энергию и относительно легко взаимодействует с последующей молекулой $Cl_2$. Энергия активации этой стадии составляет всего 2 кДж / моль. Формирование конечного соединения $CH_3Cl$ проходит через стадию второго активированного комплекса с переходным состоянием $ПС_2.$
Хлорирование метана и других алканов при таких температурах — плохоуправляемый процесс, который обычно не останавливается на стадии образования хлористого метила $CH_3C1$ и может происходить дальше:
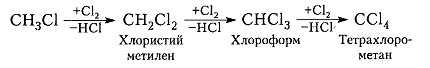
Рисунок 7. Хлорирование метана
Ионный механизм хлорирования метана
В присутствии катализаторов (кислот Льюиса $A1C1_3$, $SbF_5$), способствующих ионному механизму реакции, процесс хлорирования в кислой среде имеет электрофильный характер ($Sе$). Химизм взаимодействия електроноакцепторного хлорида алюминия с молекулой хлора заключается в значительной поляризации неполярной связи $C-C$, что вызывает её диссоциацию с образованием электрофильного агента. Без таких катализаторов реакция хлорирования по ионному механизму почти не происходит, поскольку гетеролитических расщепления молекулы хлора на катион и анион требует значительной энергии (1130 кДж / моль).
Хлорирование насыщенных углеводородов при наличии катализаторов, происходит цепным ионным електрофильным механизмом ($Se$):
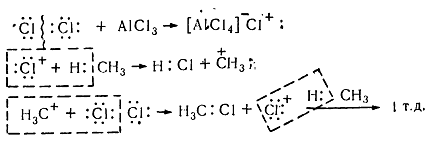
Рисунок 8. Хлорирование метана
Молекула хлора при воздействии катализатора распадается гетеролически с образованием комплексной ионной пары $[A1Cl_4]-Cl+$, поскольку алюминий соединен с электроотрицательными атомами хлора и имеет потребность в электронах. В связи с этим он отщепляет от молекулы $Cl_2$ атом хлора с парой электронов. При этом одновременно образуется катион хлора, который затем взаимодействует с молекулой метана и гетеролитично разрывает связь $C-H$. Такое взаимодействие приводит к образованию метильного карбкатиона. Последний дальше вступает в реакцию с молекулой хлора и образует хлористый метил и катион хлора, реагирует с другой молекулой метана. Такие ионные реакции, в которых промежуточными частицами являются положительно заряженные ионы, называют электрофильными.
Хлорирование насыщенных углеводородов при наличии катализаторов проводят при нагревании реакционной смеси, поскольку диссоциация молекулы хлора на ионы требует значительной затраты энергии.
Находи статьи и создавай свой список литературы по ГОСТу
Поиск по теме